CHAPITRE VI
ÉQUILIBRES ENTRE DIFFÉRENTES PHASES D'UN MÊME CORPS PUR On vient de voir dans les chapitres précédents les définitions et
les propriétés des principales fonctions thermodynamiques. Avant de les appliquer aux
systèmes chimiques et aux réactions chimiques, il convient d’en faire
l’application aux systèmes physiques et en tout premier lieu aux corps purs et à
leurs états stables. Leurs phases solides, liquide et vapeur et les passages de l’une
à l’autre, méritent une attention particulière.
Questions :
Objectifs :
1. Aspect expérimental
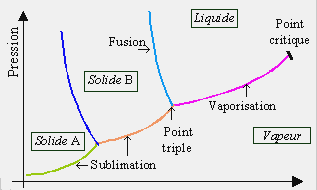
L’étude du diagramme pression-température pour un corps pur permet
d’identifier des régions où l’on ne trouvera qu’une seule phase pure (vapeur, liquide,
solide 1, solide 2, ...), des régions où coexistent deux phases en équilibre (Fig. 6.1). Ces régions sont limitées à des portions de segments curvilignes. En
appliquant la règle de la variance (voir le
Chapitre
VII.2-b), on obtient :
u
= C
+ 2 -
j Dans cette
égalité, C est le nombre de constituant : ici il n’y a qu’un seul
corps pur et j est le nombre de phases. S’il y a deux phases,
la variance u
= 1. Il n’y a qu’un seul paramètre ajustable, de telle sorte que si la pression P est fixée,
la température T l’est aussi
automatiquement et inversement. Si trois phases coexistent, la variance est nulle et
toutes les conditions de température et de pression sont déterminées par le système.
Dans le diagramme pression - température on observera un point invariant
(Fig. 6.1).
Figure 6.1. Diagramme de phase pression - température, P = ƒ(T). 2. Conditions
d’équilibre entre deux phases Soient : l’énergie molaire d’une substance pure dans les phases 1 et 2.
Généralement, ces deux quantités ont des valeurs différentes : et la phase dans laquelle la valeur de G est la plus élevée se
transforme dans l’autre phase de valeur G plus petite puisque
l’on sait que les transformations qui impliquent une variation négative de
l’énergie libre sont spontanées
(voir Chapitre
IV.3). Il existe cependant des points du
plan Pression - Température, P - T, où il y a équilibre entre deux phases. Supposons
qu’un point (Po, To)
appartienne à la région d’équilibre. Les valeurs de l’énergie libre dans chacune des
deux phases en équilibre sont égales. Supposons que ce point soit situé sur une courbe d’équilibre, de
telle sorte qu’il existe un point (Po+DPo,To+DTo) pour lequel l’équilibre existe
aussi. Lorsque la pression et la température varient, voir le
théorème d’EULER, équation [3.10],
Chapitre
III.2 : [6.3]
Þ Or le changement de phase, la transformation d’une phase 1 en une phase
2, est un processus réversible. Donc, [3.21] Dans cette équation, Équation de CLAPEYRON La courbe d’équilibre se déduit par intégration de cette équation :
la chaleur latente de changement de phase est
connue et varie avec la température. Si l’intervalle d’intégration
DT est
suffisamment grand, il faut tenir compte de la variation de la chaleur latente de
changement de phase avec la température, voir l'équation [2.18]. [6.6] Loi de KIRCHHOFF. On a déjà vu une
expression
similaire à cette loi. Cas particuliers : a) Cas de la sublimation ou de la vaporisation Puisque les phases condensées ont une densité au moins 100
fois plus grande que celle de la phase vapeur, V(phase condensée)
<< V(vapeur) [6.8] [6.9]
Si l’on intègre cette équation sur un intervalle de
température suffisamment petit,
la chaleur latente de changement de phase peut être considérée comme constante,
C’est
la formule d’YOUNG. Þ Si au contraire la variation de température,
DT, est grande, c’est-à-dire si la variation de
la capacité calorifique varie avec la
température, T : C’est la formule de KIRCHHOFF-RANKINE relative à la vaporisation de
l’eau. 3. Relations empiriques Le rapport des températures absolues pour lesquelles deux liquides A et B
ont la même pression de vapeur est approximativement constant quand P varie. Soient T'A et T'B les
températures des composés A et B ayant la même pression de vapeur P' . Soient T"A et T"B les
températures des composés A et B ayant la même pression de vapeur P" . Appliquons la loi d’YOUNG :
Dans cette équation, LA
et LB sont les chaleurs de changement de phase (ici, la vaporisation) et T'A,
T'B, les températures relatives aux composés A et B. En appliquant la loi
empirique [6.12], il vient : En d’autres termes, l’entropie de vaporisation doit être la même ou
approximativement la même pour de nombreuses substances. Si la pression est la
pression normale, P = 1 atm, l’entropie de
vaporisation, au point d’ébullition normal, est voisine de 21 unités d’entropie/mole ou
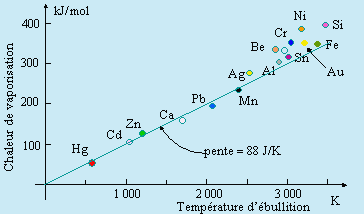
de 87,8 J·K-1·mol-1. C’est la loi de TROUTON. La figure 6.2 montre le comportement
des métaux vis-à-vis cette loi.
Il y a des exceptions notoires (Tableau 6.1). L’application de cette
loi pour les composés fortement polaires comme l’eau ou l’ammoniac doit être mise en
doute puisque pour vaporiser ces deux constituants il faut détruire une structure en
phase liquide fortement influencée par des liaisons de type dipôle-dipôle.
Figure 6.2. Les métaux et la règle de
TROUTON. Tableau 6.1. Entropies de vaporisation et loi de
TROUTON Composé Tébul (°C) DHvap
(kJ/mol) DSvap (J·K-1·mol-1)
Hélium -
268,9 100 24
Azote -
252,7 5 560 72
Butane -
1,5 22 260 82
Naphthalène -
218 40 460 82
Méthane -
161,4 9 270 83
Éther diéthylique 34,6 25 980 84
Cyclohexane 80,7 30 080 85
Benzène 80,1 30 760 87
Chloroforme 61,5 29 500 88
Ammoniac -
33,4 23 260 97
Méthanol 64,7 35 270 104
4. Considérations
supplémentaires sur les diagrammes de phase
Au voisinage du point triple, l’application de la loi de HESS
(voir Chapitre
II.5) donne
4.a Cas de la fusion L'application de l'équation de CLAPEYRON établit que :
Comme
(les volumes molaires des solides et des liquides sont
voisins), le dénominateur du membre de droite de l'équation [6.16] tend vers
zéro et le rapport
(dP/dT)fusion tend vers l’infini.
En général, le solide est légèrement plus dense que le liquide.
Donc et les courbes de fusion ont une pente verticale positive. L’eau et le
bismuth constituent deux exceptions. La glace flotte sur l’eau liquide :
Dans ces deux cas, la courbe de fusion est verticale négative.
Le
tableau 6.2 donne la température de fusion de la glace à diverses pressions.
Tableau 6.2. Variation de la température de fusion de la glace avec la
pression Pression
(atm) 1 571 1055 1490 1848 Tfus
(ºC) 0 -
5 -
10 -
15 -
20
Remarque : on détermine parfois le point de fusion sous eau. Le
composé à fondre, insoluble dans l’eau est immergé dans l’eau. Par élévation de la
température (T < 100 ºC) on obtient la fusion du composé. On a donc un système de
trois phases : le système est invariant. Ce qui est intéressant, c’est que la
température de fusion est inférieure à la température de fusion normale du composé
laissé à l’air. Cette température de fusion sous eau est appelée le point
intertectique. Par exemple, l’acide benzoïque a un point intertectique de 94 ºC alors
que sa température normale de fusion est de 121,7 ºC. Cette technique a déjà été
employée en particulier avec les médicaments sensibles à la température.
Puisque la pression a peu d’effet sur la température de fusion, une
variation mineure (disons de quelques %) de la pression sera sans effet. Cette propriété
est utilisée pour graduer les thermomètres. C’est ainsi que l’échelle internationale
Celcius sous la pression standard, atmosphérique, inclut les points de fusion
qui apparaissent dans le tableau 6.3 :
Tableau 6.3. Température de fusion de quelques métaux de
référence Éléments
Eau
Zinc
Argent
Or
Palladium
Platine Tfus
(ºC) 0,00 419,505 960,8 1063,0 1552 1769
4.b Cas de la sublimation et de la
vaporisation
Les
volumes molaires des phases condensées sont négligeables devant celui de la
phase vapeur (sauf bien sûr à des pressions élevées) :
[6.18]
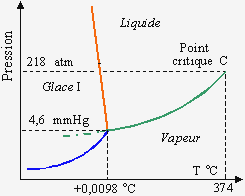
C’est évidemment surtout vrai au voisinage du point triple. Les
coordonnées du point triple dans le cas de l’eau sont : 4,581 mmHg et 0,0075 ºC
(Fig. 6.3A). Le
tableau 6.4 donne la pression de vapeur en équilibre avec la glace à diverses
températures.
Tableau 6.4. Variation de la pression de vapeur de la glace avec la
température Tsubl
(ºC) 0 -
5 -
10 -
15 -
30 -
50 Pression
(mmHg) 4,579 3,01 1,946 1,238 0,28 0,029 NOTE :
Les notions de « gaz » et de « vapeur » sont parfois confondues.
On parle bien sûr des gaz rares et de la vapeur d’eau.
Ces deux notions se rapportent à des molécules situées en phase
gazeuse. À ce titre on pourrait
croire qu’elles sont synonymes. On
trouve cependant des textes où les deux termes ont des connotations différentes.
La notion de « gaz » est alors réservée à ceux qui ont des propriétés
supérieures à leur température et pression critiques.
Ces gaz deviennent des vapeurs lorsque leurs propriétés se trouvent en
dessous des valeurs critiques. Dans
ces conditions, ces vapeurs, en général sèches, peuvent à l’occasion être
humides lorsqu’ils se trouvent en contact avec leur phase liquide.
4.c Cas de la polymorphie
C’est la propriété que peuvent avoir des solides d’exister sous
différentes formes cristallines. Chacune des structures est stable dans un domaine bien
précis de température. Le passage ou la transformation d’une phase en une autre
s’appelle une transformation allotropique. L’application de la règle des phases
à cette transformation montre que le système est monovariant : u
=
C + 2 -
j
= 1 + 2
- 2 =
1 Si donc la pression est fixée, la température l’est
automatiquement et inversement. Si la pression n’est pas un facteur à considérer,
la température de changement de phase est automatiquement fixée par le système.
C’est ainsi le cas de l’eau qui, à l’état solide peut prendre
plusieurs structures dépendant des valeurs de la pression et de la température. La
figure 6.3B localise sur le diagramme Pression - Température les régions d’équilibre. Notons
l’existence de
neuf points triples autres que celui ordinaire. Ces informations sont pertinentes à la
recherche spatiale et à l’identification éventuelle de l’eau sur d’autres
planètes. Les points triples sont situés aux coordonnées indiquées dans le tableau
6.5. L’application de la règle des phases montre que ces points sont invariants.
Figures 6.3. Différents aspects du diagrammes pression -
température, P = ƒ(T), de
l’eau.
Tableau 6.5. Les points triples de l’eau mettant en jeu au moins 2
phases solides Point Phases en équilibre * T (ºC) Pression (atm) B I III liquide -
22,0 2115 C III V liquide -
17,0 3530 E I II III -
34,7 2170 F II III V -
24,3 3510 D V VI liquide +
0,16 6380 G VI VII liquide +
81,6 22
400 H VI VII VIII ~
0 ~
20 000 I II IV
IX ~
- 100 ~
4 000 J I II
IX ~
- 100 ~
3 000 *
Les phases solides sont identifiées par des chiffres romains
Figure 6.4. Diagramme pression
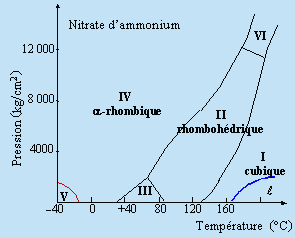
- température du nitrate d’ammonium.
Tableau 6.6. Transformations allotropiques de quelques composés Composés Changement
de phase T
(ºC)
Nitrate
d’ammonium (Fig.
6.5) tétragonal
®
b-rhombique -17 b-rhombique
®
a-rhombique 32,1 a-rhombique
®
trigonal 84,2 trigonal
®
cubique 125,2
Iodure
d’argent hexagonal
®
cubique 146,5
Nitrate
de thallium rhombique
®
trigonal 80 trigonal
®
cubique 142,5
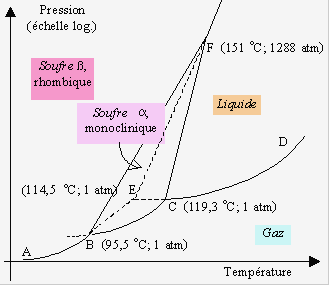
Figure 6.5. Diagramme pression-température relatif au soufre. Le soufre possède deux formes énantiotropes
a et b qui ont
chacune un domaine de stabilité bien circonscrit (Fig. 6.5). Il existe aussi des régions de ce
diagramme où l’on observe des formes métastables. Pour des raisons de vitesse de
transformation, on peut, sous la pression atmosphérique, passer directement de la phase
rhombique à la phase liquide sans passer par la phase monoclinique. La région du
triangle curviligne BCF est alors en situation métastable. On peut même observer le
point triple métastable E où coexistent (de façon métastable) le liquide, la vapeur et
la phase ß rhombique. 5. Des états particuliers
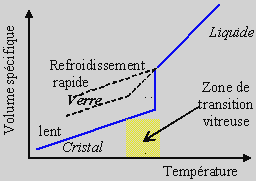
5.a Cas des verres
Il est intéressant d’introduire ici une variante importante qui donne
lieu à la formation de «verres». En effet, la silice comme plusieurs autres
oxydes de métalloïdes (B2O3, GeO2, P2O5,
As2O5 et As2O3) donne lieu à la formation de
verres qui peuvent également inclure en proportions diverses des oxydes alcalins,
alcalino-terreux et métalliques. Le principe physique de formation est simple : il repose
sur la vitesse de refroidissement du mélange en fusion. Un refroidissement suffisamment
lent permet l’édification d’un réseau périodique tridimensionnel d’atomes. Il se forme
alors de véritables cristaux. Par contre un refroidissement rapide ne le permet pas. Le
solide ainsi formé garde « la mémoire » du désordre tridimensionnel existant en
phase liquide. Par exemple, au lieu de former des structures très variées mais bien connues sous forme de feuillets ou
d’autres ensembles organisés, les tétraèdres de SiO4--
se lient au hasard avec
leurs voisins. L’agencement atomique est alors moins compact et le volume spécifique des verres est
plus grand que celui des cristaux correspondants (Fig. 6.6).
Figure 6.6. Formation d’une phase cristalline ou vitreuse à partir
d’un liquide.
5.b Cas des cristaux liquides
Certaines substances ont la propriété de former ce que l’on appelle
des cristaux liquides. Ces phases sont en quelque sorte intermédiaires en la phase
cristalline et la phase liquide puisqu’elles ont des propriétés qui appartiennent pour
certaines à la phase liquide et pour d’autres à la phase solide. Ainsi, le benzoate de
cholestéryle existe sous la phase solide jusqu’à 145,5 ºC. À cette température il a
un point de fusion plus justement appelé température de transition. La nouvelle phase
liquide quoique trouble est homogène et est anisotrope, ce qui est une propriété du
solide. Comme beaucoup de cristaux, ce liquide possède la propriété de biréfringence.
Ces cristaux liquides, ou encore cet état mésomorphe, fondent pour donner une phase
liquide à 178,5 ºC.
Les propriétés de ces cristaux liquides résultent d’arrangements
très particuliers des molécules les unes par rapport aux autres. Dans un liquide
ordinaire, les molécules sont orientées au hasard et le mouvement brownien modifie
constamment le désordre des orientations moléculaires. Dans le cas d’un liquide
nématique les molécules ont tendance à s’aligner selon leur grand axe dans une
direction privilégiée sans pour autant former de film. Le second type d’arrangement est
celui d’un liquide smectique. Il est formé par des molécules qui ont des
extrémités relativement différentes. Les parties similaires s’attirent les unes aux
autres et il y a une tendance à former des films de molécules alignées dans une
direction. Dans un troisième cas, les molécules sont alignées dans des plans (films)
parallèles dont les orientations successives se déplacent d’un angle constant.
C’est le
cas de cristaux liquides cholestériques (Fig. 6.7).
Cristaux cholestériques Figures 6.7. Arrangements moléculaires dans les
cristaux liquides.
- Quelles sont les lois qui gouvernent les changements de phases ?
- Outre les états habituels, solide, liquide et gazeuz, existent-ils
d'autres états caractérisables ?
- Comprendre les lois qui gouvernent les
changements de phases d'un corps pur
![]()
![]()
![]()
[6.1]
Donc,
![]()
[6.2]
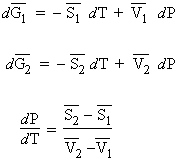
[6.4]
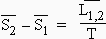
![]()
est la chaleur latente de changement de phase.
[6.5]
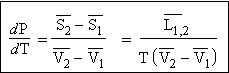
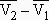
se déduit de
l’état du système. Cependant,
![]()
[6.7] Si
DT est petit :
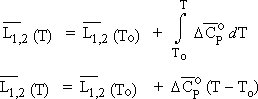
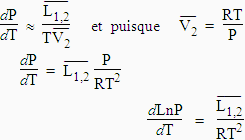
(approximation du gaz parfait)
[6.10]
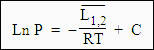
![]()
![]()
[6.11]
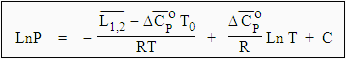
[6.12]
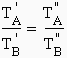
[6.13]
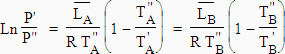
[6.14]
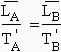
[6.15]
![]()
[6.16]
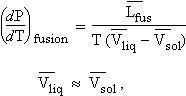
![]()
![]() .
.
[6.17]
Comme,
voir
[6.15],
A
B
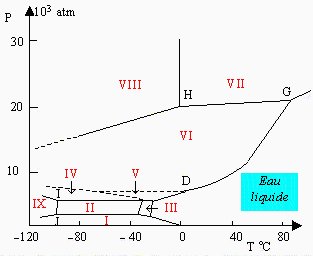
Récemment, une structure cubique à faces centrées de la glace,
structure prédite théoriquement et identifiée XVIII, a été observée à hautes
températures vers 2000 à 3000 K et à hautes pressions entre 160 et 420 GPa.
(Fig. 6.3B
et 6.3C).

![]()
Cristaux
nématiques
Cristaux
smectiques
Tableau 6.7. Quelques cristaux liquides
Constituant |
Température (ºC) de |
|
transition |
fusion du cristal |
|
Benzoate de cholestéryle |
145,5 |
178,5 |
Azoxyanisole |
118,3 |
135,9 |
Azoxyphénetol |
134,5 |
168,1 |
Acide p-méthoxycinnamique |
169 |
185 |
Bruylants, A., J. C. Jungers et J. Verhulst , Chimie générale théorique, Dunod, Paris (1961). |
||
5.c Cas des mélanges de composés énantiomères
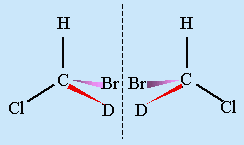
La structure tétraédrique de l’atome de carbone permet, dans le cas de substitutions appropriées, que certaines molécules organiques présentent le phénomène de stéréoisomérie. C’est aussi vrai de certains autres atomes. Les énantiomères sont des paires d’isomères qui se ressemblent comme un objet et son image dans un miroir : ils ne sont pas superposables. Ainsi, les dérivés trisubstitués ou tétrasubstitués du méthane, bi ou trisubstitués de l’azote, lorsque tous les substituants sont différents, ont cette propriété (Fig. 6.8).
 |
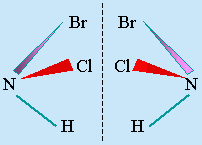 |
Figures 6.8. Images miroirs des molécules de méthane et d’ammoniac substituées.
D’autres cas d’énantiomérie découlent de la nature des liaisons,
d’empêchement stérique, ... L’étudiant trouvera dans un cours de chimie organique
approprié plus de détails sur ces composés, leur structure et leurs propriétés, en
particulier sur leur pouvoir rotatoire. Ces composés énantiomères que
l’on
représentera par la lettre ![]() et son image
miroir
et son image
miroir ![]() doivent être considérés comme
des composés différents.
doivent être considérés comme
des composés différents.
À l’état solide, le cas le plus fréquemment observé est le mélange
racémique : il est constitué de cristaux contenants chacun un mélange équimoléculaire
de composés ![]() et de son image
et de son image
![]() . Les deux composés sont présents dans la
maille élémentaire. On dit aussi que l’on a un composé racémique. Ils peuvent aussi
constituer un mélange de cristaux purs de
. Les deux composés sont présents dans la
maille élémentaire. On dit aussi que l’on a un composé racémique. Ils peuvent aussi
constituer un mélange de cristaux purs de ![]() et de
et de ![]() . Chacun des deux énantiomères est
insoluble dans la phase cristalline de l’autre. On nomme ce mélange un conglomérat.
Notons que si le mélange est un mélange 50:50, le mélange n’a aucune activité optique.
Cela n’en fait pas pour autant un mélange racémique. La fréquence d’observation de ce
type est environ dix fois plus faible que celle du cas précédent.
. Chacun des deux énantiomères est
insoluble dans la phase cristalline de l’autre. On nomme ce mélange un conglomérat.
Notons que si le mélange est un mélange 50:50, le mélange n’a aucune activité optique.
Cela n’en fait pas pour autant un mélange racémique. La fréquence d’observation de ce
type est environ dix fois plus faible que celle du cas précédent.
Ces divisions sont à l’occasion subtiles. Il arrive ainsi que certains mélanges d’énantiomères peuvent être isolés sous chacune des deux formes. C’est le cas du 1,1'-binaphthyle dont le composé racémique fond à 154 ºC et à ce moment peut se transformer en conglomérat qui, lui, fond à 159 ºC. Cette dernière forme est la plus stable à ces températures : l’autre étant dite métastable. Historiquement, c’est un conglomérat, le tartrate double d’ammonium et de sodium, qui a permis à PASTEUR (1860) de séparer mécaniquement (à l’aide d’un microscope et d’une paire de pincettes) les deux énantiomères qui produisent chacun un cristal distinct l’un de l’autre.
Il existe enfin un nombre de cas limité où la paire d’énantiomères est soluble en toutes proportions l’un dans l’autre : c’est le cas du camphre. Le mélange est alors appelé pseudoracémique. Les cas de solubilité réciproque partiels sont plus fréquents.
5.d La supraconductivité
Parallèlement à l’abaissement des températures accessibles (voir le cours de Physique moderne), on étudiait systématiquement la variation de la conductivité électrique des métaux. Le cas du mercure fut bien étudié dès 1911 par G. HOLST dans le laboratoire de K. ONNES à Leyde (Fig. 6.9). Alors que la résistance électrique diminue lentement jusque vers 4,23 K, en 3 centièmes de degrés, cette résistivité devient non mesurable (de plusieurs centaines de fois plus faibles). On montrera plus tard qu’elle est en fait nulle en dessous de 4,20 K.
|
|
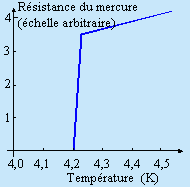
Figure 6.9. Résistivité du mercure en fonction de sa température.
L’explication théorique du phénomène fut très laborieuse : plusieurs théoriciens, des États-Unis à l’Oural, s’y attelèrent. Vers 1925, 1930 on avait établi la température critique, celle en dessous de laquelle un matériau devient supraconducteur. On avait aussi montré que cette température critique décroît lorsque le matériau est soumis à un champ magnétique croissant. Le champ magnétique appliqué et la température critique sont liés par la relation suivante :
![]()
Le tableau et la figure qui suivent explicitent cette relation et les valeurs numériques relatives pour quelques métaux. On remarquera que la zone de supraconductivité (zone située sous la demi-portion de parabole) est très limitée (Fig. 6.10).
Tableau 6.8. Effet du champ magnétique sur la température critique
Élément |
Température critique (K) |
Champ magnétique critique (tesla) |
Al |
1,19 |
0,0099 |
Pb |
7,18 |
0,0803 |
Hg |
4,15 |
0,0411 |
Sn |
3,72 |
0,0306 |
|
|
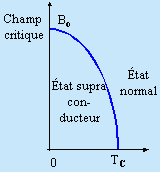
Figure 6.10. Effet du champ magnétique sur la supraconductivité.
Cette observation conduit GORTER (1934) à proposer que dans un supraconducteur le champ magnétique est nul. En 1956, trois américains BARDEEN, COOPER et J. R. SCHRIEFFER (mieux connus sous l’acronyme BCS) proposent une explication théorique satisfaisante de la supraconductivité. Cependant cette théorie n’a aucune valeur prédictive de telle sorte que depuis que le phénomène est connu, les expérimentateurs sont laissés à eux-mêmes quant aux matériaux à synthétiser pour amener la supraconductivité vers des températures accessibles. En 1954, un allemand émigré aux États-Unis, B. MATTHIAS, découvre qu’un alliage de formule Nb3Sn montre une température critique d’apparition de la supraconductivité de 18,5 K soit beaucoup plus que les quelques degrés jusqu’alors observés. Il se lance dans un vaste programme d’étude des alliages. Il montre que plusieurs de ceux-ci dont la valence se situe autour de 5 sont supraconducteurs : le meilleur d’entre eux, Nb3(Al2Ge) est supraconducteur à 21 K (Fig. 6.11).
|
|
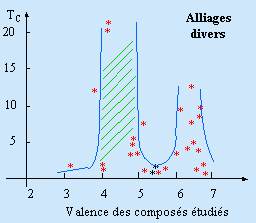
Figure 6.11. Température critique de divers alliages étudiés par
MATTHIAS.
(Note : la zone hachurée représente une cinquantaine de mesures
expérimentales).
L’étude systématique de la supraconductivité des matériaux nouvellement synthétisés par les chimistes minéralistes a connu ses heures de gloire en France et aux États-Unis en passant par l’Extrême-Orient et la Suisse. En 1971, le PbMo6S8 montre une supraconductivité au-dessus de 14,4 K avec un champ magnétique critique de 60 teslas. Les essais du côté des composés organiques et des polymères auront assez peu de succès. Des chercheurs suisses (laboratoires d’IBM) vont ouvrir un nouveau champ d’exploration en observant que certains oxydes présentent cette propriétés. Les composés du type BaPb(1 - x)BixO3 sont supraconducteurs (Tmax. 13 K). Ces composés ont la structure des pérovskites naturelles ou synthétiques : formule ABX3 (exemple : CaTiO3). Vers la fin des années 80, les recherches s’orientent vers des composés de type Ba-La-Cu-O, puis Ba-Yb-Cu-O, puis Y-Ba-Cu-O, ... (Tableau 6.7). Plus récemment, on a eu la surprise de voir des composés tels que le borure de magnésium, MgB2, ainsi que des composés dans lesquels des atomes de fer sont présents, particulièrement des matériaux de type Fe-As dopés avec des atomes de fluor, - exemple : LaO0,89F0,11FeAs - avoir des propriétés supraconductrices suggérant un lien entre le magnétisme et la supraconductivité. Ces découvertes ont produit un regain d'activités sur les supraconducteurs.
Tableau 6.7. Progression récente des températures critiques observées
Année |
Composé |
Température critique |
1986 |
La1,8Sr0,2CuO4 |
38 K |
1987 |
YBa2Cu3O7-e |
93 K |
1988 |
(Bi ou Pb)2Sr2Ca2Cu3O10-e |
110 K |
1991 |
Tl1,6Ba2Ca2Cu3O10-e |
128 K |
1993 |
HgBa2Ca2Cu3O8-e |
138 K |
note : e << 1. |
||
Si les cuprates (tableau précédent) semblent importants en ce qui concerne la supraconductivité, on a rapporté les mêmes propriétés avec le borure de magnésium, MgB2, à 39 K ainsi qu’avec le LnN2B2C3 à 17 K. Les fullérènes et les nanomatériaux ne cessent d’étonner par leur structure et leurs propriétés. Certains fullérènes sont supraconducteurs à 50 K. Les découvertes restent donc à venir.
Il faut rappeler l’importance de la recherche de la supraconductivité.
Bien sûr, la première application qui vient à l’esprit elle celle du transport de
l’énergie électrique sur des grandes distances afin d’éviter les pertes par effet
joule. Tant que l’on n'aura pas mis au point un matériau ayant une température critique
voisine de la température ordinaire, les coûts de refroidissement, même avec de la
neige carbonique, seront beaucoup plus élevés que ceux encourus par l’utilisation
d’une
ligne à très haute tension comme celles développées par Hydro-Québec sous 735 kV. En
ce qui a trait à la fabrication d’électroaimants à haute performance, on pense ici à
la lévitation magnétique et à la mise en œuvre de trains à très haute vitesse
sur support magnétique, on retrouve les mêmes difficultés. Les Japonais ont construit
et testé un train mais à quel prix! À certaine époque on considérait qu’on ne saurait
jamais faire circuler un train à plus de 200 km/h. Le TGV français a roulé à plus de
500 km/h et à plus de 300 km/h en fonctionnement commercial. La supraconductivité
demeure pour le moment limitée à des applications de laboratoire.
5.e
Le magnétisme On a déjà vu au chapitre I la
transformation du magnétisme du fer a
révélée par la variation de capacité calorifique vers 1033 K (Fig.
1.3). En
réalité il existe plusieurs formes de magnétisme selon l’arrangement des
vecteurs de moments magnétiques atomiques. On observe ainsi des solides
paramagnétiques (aucun arrangement de ces vecteurs) ferromagnétiques,
ferrimagnétiques et antiferromagnétiques (orientations particulières de ces
vecteurs) (voir le cours "Chimie théorique",
Chapitre
XVII.4). On trouvera des informations plus précises dans des cours
appropriés. Disons seulement ici que, dans ces cas, on observe des domaines de
températures où ces propriétés existent et d’autres régions où elles n’existent pas. On a donc des régions de changement de magnétisme tout comme
il existe des régions de changement de phase. Les corps purs peuvent exister sous la forme de diverses phases
solides, sous la forme liquide et sous la forme gazeuse. Chaque région du diagramme
Pression - Température est séparée par des courbes de changement de phase qui ont la particularité de se
rejoindre trois par trois en un point triple invariant. Les formules d’YOUNG et de
KIRCHOFF-RANKINE sont des exemples d’équation de ces courbes. Un corps pur peut exister sous différentes formes solides (cas de la
polymorphie). Certains de ces solides présentent des propriétés particulières de
supraconductivité à basse température. Finalement, dans des cas particuliers, la courbe
de fusion laisse place à des phénomènes caractérisés par des cristaux liquides,
formes condensées intermédiaires entre la forme solide et la forme liquide. 7.1. Le tableau suivant donne les résultats des mesures sur le point
de fusion de la glace en fonction de la pression ainsi que les variations de volume
observé lors des changements de phase. Calculez P
(atm) T
(° C) DV (cm3/kg) 1 590 1090 1540 1910 0,0 -
5,0 -
10,0 -
15,0 -
20,0 -
90,0 -
101,6 -
112,2 -
121,8 -
131,3 7.2. Entre -
75 et + 125 ºC, la pression de vapeur du 1-butène en
fonction de la température est la suivante : Calculer : 1- la variation d’enthalpie
DH de vaporisation en fonction de la
température T ; Réponses : la variation d’enthalpie,
DH, de vaporisation en fonction de la température
est donnée par la relation :
25,6 -3,19 10-5
T2 kJ mol-1. 2- la valeur de DH à 300 K ; 3- la température d’ébullition du 1-butène.
T1-butène
= 267,3 K.
7.3. Les pressions de vapeur de l’arsenic liquide et gazeux sont
données par les équations : Cas du
liquide : log10 P(mmHg) = -2 460 / T +6,99 Cas du solide : log10
P(mmHg) = -6 947 / T +10,8 Trouver la température à laquelle l’arsénique solide et
l’arsenic
liquide ont la même pression de vapeur. Trouver également cette pression. Que
pensez-vous de ce point ? Réponse : P =
104,5 atm.
7.4. Les variations de l’enthalpie et d’entropie relatives à la
sublimation du cuivre, Cu(s) ®
Cu(g), sont données par les relations
suivantes : DHo = 81 730 -0,47 T -0,731 10-3 T2, et DSo = 34,94
-1,08 log10
T -1,46 10-3
T. Établissez l’expression de
DGo en fonction de la
température T. Réponse : DGo =
81 730 -35,41 T
+ 2,19 10-3 T2 + 0,47 T Ln T
7.5. La pression de vapeur du cadmium métallique est
de 10-5 et de 10-4 mmHg respectivement à 148 et à 180
ºC. Calculez la chaleur latente de sublimation
du cadmium au voisinage de 165 ºC.
Réponse
: la chaleur latente de sublimation
du cadmium est de 120,36
kJ mol-1. 7.6. La masse molaire de l’eau est de 18 g. La densité de la glace est de 0,917 g/
cm3. En déduire au voisinage de 0 ºC la valeur de la tangente à la courbe P =
¦ (T) représentant la courbe de fusion de la glace. On indique que
la chaleur latente de fusion de la glace est de 335 J/g. Réponse : dP
/ dT
=
3,08 J · °C · mol-1. 7.7. La température d’ébullition sous la pression
atmosphérique de la 2-hexanone est de 127,5 °C. À la
pression de 100 mmHg, cette cétone bout à 79,8 °C. La température d’ébullition sous la pression atmosphérique de la
2-heptanone est de 150,2 °C. Quelle sera sa
température d’ébullition sous une pression de 100 mm de Hg ? Réponse : T "B
= 99,79 °C.
7.8. En admettant que l’enthalpie de vaporisation de
l’eau est de 41 400 J/mol, calculer la température à laquelle la pression de
vapeur saturante de l’eau est de 2 atmosphères. Réponse :
T = 120,25 °C. 7.9. On détermine la tension de vapeur du
tétrachlorure de carbone, CCl4, liquide en fonction de la température. Si la
pression P est exprimée en mm de mercure, on obtient l’équation suivante :
Calculez la chaleur latente de
vaporisation du CCl4 à 77 °C. Réponse : DHvap
=
30,4 kJ/mol.
Pour en savoir un peu plus Eliel, E. L. et S. H. Wilen, Stéréochimie des composés
organiques, traduction : R. Panico et Richer, J.-Cl., Lavoisier TEC-DOC, Paris
(1996). Matricon, J. & Georges Waysand, La guerre du froid, Éd.
du Seuil, Paris, 1994. B. Raveau et Cl. Michel, Les supraconducteurs à
haute température critique, L’actualité chimique, 150-156,
mars 2002. On trouvera aussi quelques
diagrammes pression température de composés pur sur
le Net. S. Lemonick, Scientists make
superionic ice,
Chemical & Engineering News, 6 (20 mai 2019). Les verres
ophtalmiques et plus particulièrement les lentilles cornéennes sont à la
mode. On trouvera des éléments intéressants à leur sujet dans la revue
de la Société chimique de France : Pierlot, C, A. Vanrobaeys et A. Colonna de
Lega, L'Actualité chimique, 279, 8-14 (octobre 2004).
Également sur le web, et en provenance de l'ESPCI, Paris, Sophie Norvez,
Les cristaux liquides - D’un état insoupçonné de la matière aux écrans plats,
https://cours.espci.fr/site.php?id=57&fileid=1466 (visité le 2019-03-03). On trouvera le diagramme pression - température du carbone
ainsi que beaucoup d'informations dans la revue de la Société chimique de
France (page 118) : « Les matériaux carbonés :
recherche et applications », L'actualité chimique (Société chimique de France), N° 295-296, mars-avril 2006. Les derniers développements sur la supraconductivité, voir
ou lire : Des sites web très pertinents ou intéressants, aussi sur Wikipedia : BARDEEN,
COOPER, SCHRIEFFER :
https://www.nobelprize.org/prizes/physics/1972/summary/
Dernière mise à jour : 2021-07-08. x x x x x x x x x x x x x
![]()
![]()
M. Jacoby, Superconductivity rekindles, Chemical & Engineering News,
15-21 (octobre 2008).
ONNES :
http://www.nobelprize.org/nobel_prizes/physics/laureates/1913/onnes-bio.html