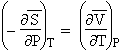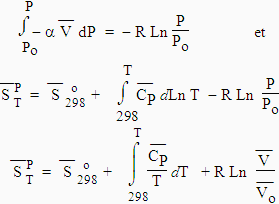[3.1] |
|
CHAPITRE III
LES 2ème ET 3e PRINCIPES DE LA THERMODYNAMIQUE
|
On a vu que chaleur et travail sont deux formes d’énergie qu’un système peut échanger avec l’extérieur. Questions : Objectifs : |
1. Rappels généraux
Deuxième principe :
Pour qu’un système évolue de manière réversible il faut que son entropie soit constante, ou encore que la variation de cette dernière soit nulle, DS = 0. Pour un système qui évolue irréversiblement, DS doit être nécessairement positif. La conséquence est que l’entropie de l’univers croît ou est maximum. Elle serait maximum si l’univers était figé. Comme ce n’est pas le cas elle croît.
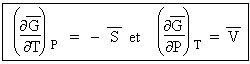
Pour un système quelconque, on peut toujours transformer du travail en chaleur. L’inverse n’est pas vrai et le produit TDS mesure la quantité de chaleur non transformable en travail. L’énergie utilisable G est alors telle que :
[3.1] |
|
L'énergie utilisable est égale à l'enthalpie diminuée du produit température × entropie. Comme les valeurs absolues de ces fonctions énergie libre et enthalpie ne sont pas accessibles, on écrit plutôt :
| [3.2] |
|
On mesure ainsi les variations de ces fonctions au cours d’une transformation.
2. Variations de l’entropie avec la température et la pression
La variation de l’énergie utilisable G est telle que donnée par la relation [3.3] (Note : G = f(T, P)) et l’équation [3.1] se dérive selon la relation [3.4] :
| [3.3] [3.4] |
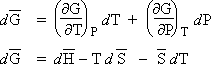 |
ou encore |
À partir de la relations [2.3], on peut réécrire l’équation précédente :
| [3.5] | car : H = E + PV |
et en se rappelant que q est la quantité de chaleur échangée avec l'extérieur, voir Chapitre 2.4, relation [2.2],
[3.6] dE = q - w .
Dans le cas d’une réaction réversible (voir un premier cours de thermodynamique) :
[3.7] q = T dS et w = P dV
Note : dans le cas d'une transformation irréversible : T dS > q et P dV > w.
| [3.8]
|
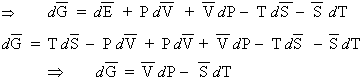 |
D’où le théorème d’EULER :
| [3.10] |
|
Puisque la dérivée de la fonction G par rapport à la température T suivie de la dérivée du résultat par rapport à la pression P est égale à la dérivée de la même fonction par rapport à P suivie de la dérivée du résultat par rapport à T, autrement dit, puisque l’ordre des dérivées peut être permuté (c'est l'expression de la différentielle exacte) :
| [3.11] |
En remplaçant les valeurs obtenues dans l'équation [3.10], on obtient :
| [3.12] |
|
Relation de MAXWELL. |
Puisque par définition, le coefficient d’expansion volumique, a, est tel que :
|
|
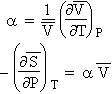 |
On peut aussi montrer que
| [3.14] | 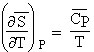 |
de telle sorte que si la fonction entropie dépende de la température et de la pression, on obtient :
| [3.15]
[3.16] Þ |
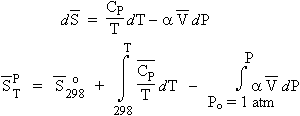 |
a) Cas des liquides et des solides supposés incompressibles :
Comme il n’y a pas de variation de volume, le travail contre les forces de pression est nul :
| [3.17] Þ | 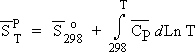 |
car |
est négligeable; ou bien encore a » 0. |
b) Cas des gaz parfaits :
|
|
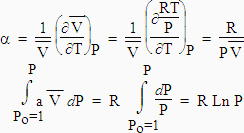 |
En général, si la pression est différente de la pression atmosphérique, P ¹ 1 atmosphère,
| [3.18]
ou [3.20] |
|
puisque
. (C’est la loi de BOYLE-MARIOTTE).
3. Entropie et changement d’état
Lorsqu’ils se font naturellement, les changements d’état ou de phase sont des processus réversibles. Par conséquent, la variation d’entropie est égale au rapport de la variation d’enthalpie sur la température de changement d’état :
| [3.21] |
Dans cette équation
|
|
est la chaleur latente de changement de phase. |
Exemple 1 : Soit la transformation H2O (s) ® H2O (liquide) à 273 K.
| La valeur de |
|
= 80 cal·g-1 = 1440 cal·mol-1 = 6019,2 J·mol-1 |
| et celle de |
|
= 22,05 J·K-1·mol-1 |
Rappel : 1 calorie est l’équivalent de 4,18 joules.
Exemple 2 : Soit la réaction S(monocl.) ® S(rhomb.)
On donne les valeurs des capacités calorifiques molaires entre 250 et 368,5 K :
| (S, monocl.) = 3,493 + 0,006 36 T cal·K-1·mol-1 | |
| (S, rhomb.) = 3,556 + 0,006 98 T cal·K-1·mol-1 |
Calculer l’entropie de la réaction en fonction de la température,
|
= ƒ(T). |
L’application de la relation [3.17] conduit à :
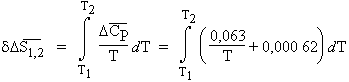
puisque
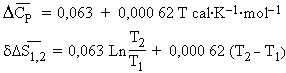
La connaissance de
![]()
à une température permet de connaître les valeurs correspondantes à n’importe quelle température pourvu que l’on connaisse les variations des capacités calorifiques.
Cas de la vaporisation.
Une règle empirique intéressante a été proposée par F. TROUTON en 1884 (voir plus loin, Chapitre VI.3). En règle générale,
La chaleur de vaporisation, exprimée en joules par mole, divisée par la température d’ébullition exprimée dans l’échelle KELVIN, ou encore l’entropie de vaporisation, est approximativement égale à 88 J·K-1·mol-1. |
Le tableau 3.1 donne quelques valeurs numériques relatives à cette loi. La dernière colonne du tableau permet d’apprécier la validité de cette loi ainsi que des exemples de molécules qui s’écartent de cette loi.
Tableau 3.1. Enthalpie et entropie de vaporisation de quelques molécules
Molécules |
T(ébull.) |
|
|
Butane |
271,5 |
22 260 |
82,0 |
Naphtalène |
491 |
40 460 |
82,4 |
Méthane |
111,6 |
9 270 |
83,1 |
Éther diéthylique |
307,6 |
25 980 |
84,5 |
Cyclohexane |
353,7 |
30 080 |
85,0 |
CCl4 |
349,7 |
30 000 |
85,8 |
SnCl4 |
385,7 |
33 050 |
85,7 |
Benzène |
353,1 |
30 760 |
87,1 |
Trichlorométhane |
334,5 |
29 500 |
88,2 |
Hydrogène sulfuré |
213,4 |
18 800 |
88,1 |
Mercure |
629,6 |
59 720 |
94,1 |
Ammoniac |
239,6 |
23 260 |
97,1 |
Voir Barrow, G. M., Chimie Physique, tome 2, Masson, (1976). |
|||
4. Le 3e principe
(ou la 3e loi)
Par définition, on doit admettre que l’entropie de toute
substance cristallisable est nulle à la température du zéro absolu. On peut donc déterminer les valeurs de l’entropie standard à 298 K en calculant
l’intégrale suivante : La mesure de l’entropie d’une substance revient à mesurer la
capacité calorifique à pression constante entre 0 et 298 K. Il ne faut pas oublier en
cours de route de tenir compte des changements éventuels de phase. 5. La table d’entropie
[3.22]
![]()
à la température du
zéro absolu : T = 0 K.
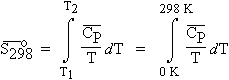
Tout comme pour l’enthalpie, on trouvera des tables donnant les
entropies de diverses substances dans les conditions standards. Ces tables sont
déterminées de plusieurs façons. Bien entendu, l’une de ces façons consiste à
utiliser le troisième principe ci haut énoncé et de faire, par exemple, l’intégration
graphique entre la température du zéro absolu et 298 K de la fonction ![]() /T . Une
autre façon utilise les mouvements de translation, rotation, vibration, ... auxquels on
applique la fonction de partition. L’étudiant verra (ou a déjà vu) cette méthode dans
un cours de chimie théorique (voir particulièrement le
Chapitre II).
/T . Une
autre façon utilise les mouvements de translation, rotation, vibration, ... auxquels on
applique la fonction de partition. L’étudiant verra (ou a déjà vu) cette méthode dans
un cours de chimie théorique (voir particulièrement le
Chapitre II).
On doit aussi rappeler, comme ce fut le cas de l’enthalpie, les variations d’entropie de réactions chimiques semblables doivent être semblables. Ainsi les réactions d’oxydation partielle des hydrocarbures saturés en monoalcools demandent les mêmes réactions de rupture et de reconstitution de liaisons quelle que soit la nature de l’alcane initial (Tableau 3.2) :
Tableau 3.2. Entropie de combustion partielle de quelques molécules
Réaction |
DS (J·K-1·mol-1) |
CH4 + 1/2 O2 ® CH3OH |
- 48,5 |
C2H6 + 1/2 O2 ® C2H5OH |
- 49,5 |
C3H8 + 1/2 O2 ® n-C3H7OH |
- 47,5 |
n-C6H14 + 1/2 O2 ® n-C6H13OH |
- 48,5 |
6. Conclusions
La transformation complète ou maximale chaleur - travail ne peut se réaliser que dans des conditions réversibles, ce qui implique que le travail réalisé est nul. Dans les cas concrets de transformation, la différence entre la chaleur disponible et le travail réalisé se mesure par le produit T DS où DS représente la variation d’entropie du système. Cette fonction entropie est reliée au rapport CP/T et peut se calculer théoriquement pourvu que, par définition, l’entropie de toute substance est nulle au zéro absolu. |
7.1 Calculer la variation d’entropie subie par 2 moles d’un gaz qui passent d’un volume de 100 litres à 50 °C à un volume de 150 litres à 150 °C. On indique que dans les présentes conditions, la capacité calorifique à volume constant de ce gaz est de 33 J/(mol· degré).
7.2 La capacité calorifique du triptycène (C20H14) est déterminée à différentes températures (Tableau suivant).
|
En déduire son entropie |
|
à l’état solide |
|
ainsi que la valeur |
|
du composé liquide en J·(K·mol)-1. |
Nota : Entre 0 et 10 K, on utilisera la loi de DEBYE
|
= a T3 |
et on fera l’intégration graphique nécessaire au-dessus de 10 K.
Capacité calorifique (cal·(K·mol)-1) molaire du triptycène
T (K |
10 |
20 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
(s) |
0,863 |
4,30 |
7,73 |
10,65 |
13,17 |
15,4 |
17,4 |
19,3 |
21,2 |
T (K |
100 |
120 |
140 |
160 |
180 |
200 |
220 |
240 |
260 |
|
23,0 |
26,7 |
30,55 |
34,6 |
38,9 |
43,4 |
48,0 |
52,8 |
57,8 |
T (K |
280 |
298,15 |
320 |
350 |
400 |
450 |
500 |
527,18 |
|
|
62,9 |
67,6 |
73,2 |
80,7 |
92,5 |
103,8 |
114 |
119,4 |
|
T (K |
527,18 |
530 |
550 |
||||||
|
130,8 |
130,9 |
133,4 |
Le composé fond à 527,18 K avec une chaleur latente molaire de 53,67 kJ·mol-1.
Réponse : en utilisant de manière appropriée, le tableur Excel, on obtient :
S° (trypticène) = 270,59 J/(mol·K) à 298 K
S° (trypticène) = 637,18 J/(mol·K) à 550 K.
Dernière mise à jour : 2021-07-08.
X X X X X X X X X X X X X