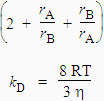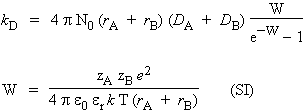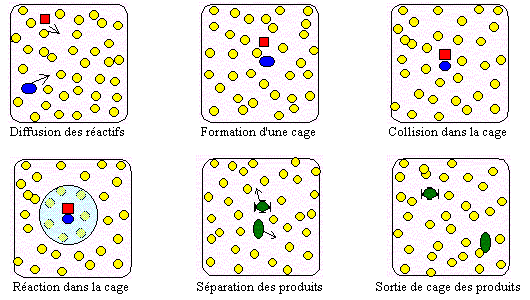
Figure 9.1. Schématisation d’une réaction dans un solvant inerte.
CHAPITRE 9
LA CINÉTIQUE EN PHASES CONDENSÉES
Préambule/Objectifs L’étudiant aura remarqué que, jusqu’ici, on a surtout traité de réactions ayant lieu en phase gazeuse, là où le mouvement brownien est le support des collisions physiques et où les molécules ont toute liberté de se mouvoir au sein du milieu réactionnel. La phase condensée vient limiter de manière importante cette liberté de mouvement.
|
Objectifs spécifiques
- Assimiler les conséquences de l’immersion de réactifs chimiques dans des phases condensées sur les mécanismes et les vitesses de réaction.
- Comprendre les effets de viscosité ou de diffusion, de cage, de force ionique sur la vitesse de réaction.
- Étudier l’effet de la pression sur les réactions dans un solvant ou en solution.
- être capable de maîtriser la notion de volume d’activation.
On a plus spécifiquement étudié les réactions en phase gazeuse en y appliquant plus particulièrement la théorie cinétique des collisions. L’ensemble des relations qui en découlent sont encore valables en phases condensées. Il faut bien sûr y ajouter un certain nombre de considérations nouvelles. Parmi ces éléments il faut tenir compte d’une fréquence de collisions beaucoup plus élevée. Mais ce n’est pas de cela que viendra la principale différence. Alors qu’en phase gazeuse et à la pression atmosphérique, on peut considérer que les molécules sont isolées, sans influence mutuelle, ce n’est souvent plus le cas en phase condensée. Rappelons que si, en phase condensée, la densité est environ 1 000 fois plus grande qu’en milieu gazeux, à la pression atmosphérique, la distance intermoléculaire moyenne y est 10 fois plus faible. Ces interférences seront particulièrement importantes dans des milieux contenant des molécules polaires ou même des solvants ionisants. On verra aussi que dans le cas de milieu à viscosité élevée, des problèmes de transport de matériaux viendront interférer avec le bon déroulement réactionnel. Enfin, comment peut-on, tout en gardant le milieu homogène, provoquer des réactions en phase solide ?
L’effet du solvant sur les constantes de vitesse n’est pas a priori évident. Dans le cas de réactions bimoléculaires mettant en jeu des ions, ceux-ci sont souvent solvatés. Ils traînent donc avec eux un cortège de molécules qui les alourdissent et modifient leur champ électrique. Dans le cas où les molécules sont polaires ou ioniques, la constante diélectrique, er, du milieu (solvant) intervient.
Tableaux 9.1. Exemples de réactions ioniques
Réaction : CH3I + Cl- ® CH3Cl + I- (*)
| Solvant | HC(=O)NH2 | HC(=O)NHCH3 | HC(=O)N(CH3)2 |
| k(dm3·mol-1·s-1) | 5 10-5 | 1,4 10-4 | 4 10-1 |
| (*): cette
réaction fait l'objet d'une étude spectaculaire qui en a
décortiqué le mécanisme. Voir ce cours, chapitre 2.8 ainsi que la revue Science, 319, 183 (2008). |
|||
Réaction : C2H5I + N(C2H5)3 ® [N(C2H5)4]+ I-
| Solvant | er | k (relatif) à 100 °C |
| hexane benzène chlorobenzène acétone nitrobenzène |
1,88 2,27 5,63 20,7 34,8 |
1 80 332 530 2766 |
Dans le second cas, il existe une corrélation au moins apparente entre la constante diélectrique er du solvant et la constante de vitesse de la réaction. Par contre, pour des réactions bimoléculaires ne mettant pas en jeu des espèces chargées, la constante ne dépend pas ou très peu du solvant :
Tableau 9.2. Réaction de dimérisation du cyclopentadiène : C5H6
| Milieu | gaz | CS2 | C6H6 | C2H5OH | CCl4 | paraffine | CH3COOH |
| k(dm3·mol-1·s-1) | 6,2 10-6 | 7 10-6 | 10 10-6 | 20 10-6 | 16 10-6 | 25 10-6 | 22 10-6 |
Lorsqu’un réactif est dilué dans un solvant, plusieurs effets peuvent apparaître. L’effet de cage est le plus simple et peut se visualiser assez simplement. On imagine, en effet assez facilement que le solvant exerce une barrière physique vis-à-vis la diffusion des produits réactionnels. Cela est d’autant plus évident que le solvant est visqueux, voire même solide.
Soit, par exemple, la photolyse de l’azométhane en phase gazeuse. C’est une réaction de décomposition bien connue :
[1] CH3N=NCH3 + hn
® 2 CH3•
+ N2
En mélangeant l’azométhane à son semblable perdeutérié (mélange 50-50), il y aura formation de radicaux méthyles eux aussi perdeutériés. Ces radicaux se combinent entre eux pour former l’éthane-d0, -d3 et -d6 :
| [2] | CD3-N=N-CD3 + hn | ® | 2 CD3• + N2 |
| [3] | •CH3 + •CH3 | ® | CH3-CH3 |
| [4] | •CH3 + •CD3 | ® | CH3-CD3 |
| [5] | •CD3 + •CD3 | ® | CD3-CD3 |
Si l’on réalise la même photodécomposition en solution, dans l’isooctane, on ne trouve que la formation de l’éthane-d0 et -d6. On doit donc admettre que l’isooctane maintient les produits de photodécomposition dans un volume extrêmement réduit de telle sorte que, la diffusion n’ayant pas lieu, les radicaux méthyles provenant de la même molécule d’azométhane sont les seuls à pouvoir se recombiner. On a observé le même effet dans la radiolyse du néopentane, (CH3)4C, liquide.
Cet effet a été utilisé pour synthétiser et piéger des espèces autrement très instables. Soit une matrice solide d’azote (T < - 215 °C) contenant des traces d’oxygène moléculaire et d’iodure d’hydrogène soumis à un rayonnement du proche UV. Seule la molécule HI absorbe la lumière : il s’ensuit une décomposition photochimique en hydrogène et iode atomiques. L’atome d’hydrogène est petit et léger de telle sorte qu’il s’échappe facilement à travers le réseau cristallin d’azote et finit par être intercepté par l’oxygène moléculaire. L’atome d’iode, au contraire, reste piégé à la même place dans la matrice solide. On forme ainsi des radicaux HO2• piégés dans le réseau solide. On a pu de cette façon mesurer le spectre infrarouge d’une espèce autrement très fugace.
[6] HI + hn
® H•
+ I•
[7] H• + O2
® HO2•
4. L'effet de diffusion (ou effet de viscosité)
1- Interaction entre des particules neutres
Soient deux réactifs A et B en solution (molécules de solvant inertes vis-à-vis A ou B) conduisant à la formation de produits M et N.
Figure 9.1. Schématisation d’une réaction dans un solvant inerte.
En tout premier lieu, les réactifs A et B doivent diffuser dans le liquide pour éventuellement se retrouver dans une cage constituée de molécules de solvant. Ils accumulent et perdent de l’énergie par collision avec les molécules de solvant et forment éventuellement un complexe réactionnel (A-B) avec une constante de vitesse kD. Le complexe peut se redécomposer, constante de vitesse k-D, ou former les produits avec une constante de vitesse kr. En appliquant le principe de quasi-stationnarité au complexe, il vient (pour plus de détails, voir le traitement réalisé sur la catalyse enzymatique) :
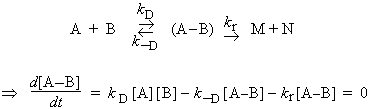
La concentration du complexe intermédiaire devient :
![]()
La vitesse
de formation des produits M et N est donnée par :
|
v = kr [(A-B)] = |
et vexp = kexp [A] [B].
La constante de vitesse expérimentale est égale à :
![]()
La réaction est d’ordre 2.
Deux cas limites deviennent évidents.
![]()
La vitesse de réaction est intimement liée à la constante thermodynamique de l’équilibre, KD, ainsi qu’à la vitesse de passage de la barrière de potentiel située entre le complexe (A-B) et les produits M et N.
v = kD [A] [B]
La vitesse de réaction est contrôlée par la réaction de formation du complexe. La vitesse de réaction est donc limitée par la vitesse de diffusion des réactifs dans la solution. Par exemple, dans l’eau à 25 ºC, kD est voisin de 109 - 1010 litre·mol-1·s-1.
À viscosité infinie, le cas limite correspondant sera l’effet cage précédemment vu. SMOLUCHOWSKI, en 1917, a montré que la constante de vitesse de diffusion, ou encore mieux appelée la vitesse de rencontre, kD, d’une particule est telle que
9.1 kD = 4 p N (rA + rB) (DA + DB)
DA et DB sont les coefficients de diffusion de A et de B dans le solvant considéré
N est le nombre d’AVOGADRO, et
rA et rB sont les rayons des particules A et B dans le solvant utilisé
Dans le cas où A et B sont identiques, l’équation devient :
kD = 2 p N (2 rA) (2 DA) = 8 p N rA DA
Il faut noter que la constante de "rencontre", kD, dépend directement du rayon de collision (rA + rB) alors que la constante de vitesse de collisions physiques dépend du carré de cette même quantité : voir Chapitre 2. Expérimentalement, on peut vérifier la validité de cette loi. Bien que l’accord entre les valeurs observées et mesurées ne soit pas parfait, il est cependant suffisant pour vérifier la validité de l’approche. Enfin, bien que le dernier exemple du tableau 9.3 implique une interaction ion-dipôle, la très grande valeur de la constante diélectrique de l’eau réduit très fortement la contribution du potentiel électrostatique à la constante de vitesse.
L’équation 9.1 peut être simplifiée en tenant compte de l’équation de STOKES-EINSTEIN reliant la diffusion d’une sphère D à la viscosité, h, du milieu à la température T :
| équation de STOKES-EINSTEIN : |
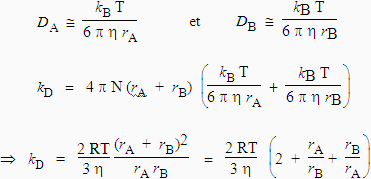 |
En posant : rA / rB = 1 + a et rB / rA = 1 - a, (note : rA @ rB)
|
Þ |
|
= 2 + (1 + a) + ( 1 - a) = 4 |
| dans le cas où A ¹ B, non-ioniques |
et
| 9.2 Þ |
|
dans le cas où A = B, non-ioniques |
Le facteur 2 qui existe entre les deux dernières équations tient à un seul problème de statistique. Dans le cas où A et B sont identiques, la fréquence de collision entre deux particules A est deux fois plus élevée que dans un système où la moitié des molécules sont A et l’autre moitié sont B.
Tableau 9.3. Comparaison entre les vitesses de diffusion mesurées et calculées
| Réaction | Solvant | T | DA et DB | rA + rB | k × 10 M-1 (1/s) | ||
(°C) |
(cm-2 ·s-1) |
(nm) |
calc. 9.2 |
observée |
|||
| I• + I• | ® I2 | CCl4 | 25 | 2 | 0,4 | 1,3 | 0,82 |
| •OH + •OH | ® H2O2 | H2O | 24 | 2,6 | 0,3 | 0,6 | 0,5 |
| •OH + C6H6 | ® C6H5OH | H2O | 24 | 2,6-1,1 | 0,3 | 0,8 | 0,33 |
| •C2H5 + •C2H5 | ® n-C4H10 | C2H6 | -177 | 0,7 | 0,2 | 0,1 | 0,02 |
| H+ + NH3 | ® NH4+ | H2O | 25 | 10-3 | 0,4 | 4,0 | 4,3 |
Note : DA et DB sont les coefficients de diffusion des espèces en réaction.
2- Interaction entre des particules chargées
Si A et B sont des espèces chargées, l’attraction ou la répulsion électrostatique jouant, l’équation 9.1 doit être corrigée pour tenir compte de ces forces (DEBYE, 1942) :
| 9.3 |
|
|
où |
Les variables zA et zB sont les nombres de charges électriques portées par les particules A et B et k est la constante de BOLTZMANN.
Dans le système CGS, la constante de vitesse de diffusion peut s’exprimer sous la forme :
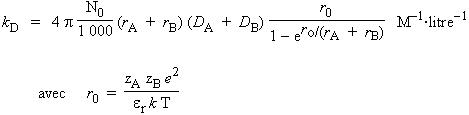
La grandeur e est la charge électrique exprimée en unité CGS-UES : e = 4,8 10-10. Selon la théorie de DEBYE-HÜCKEL, pour l’eau er = 78,4 et pour des rayons de 0,5 nm et à 25 °C, les valeurs W/(e-W - 1) sont données dans le tableau 9.4. Comme on peut le voir, deux ions de même signe seront répulsifs l’un envers l’autre et le facteur W/(e-W - 1) est inférieur à l’unité. Inversement, l’attraction électrostatique de deux ions de signe contraire augmentera la vitesse de diffusion et le facteur est supérieur à l’unité.
Tableau 9.4. Valeurs de W/(e-W - 1) dans le cas d’ions dans l’eau
| zA,zB | 1,1 | 2,1 | 2,2 | 1,-1 | 2,-1 | 2,-2 | 3,-1 |
| W/(e-W - 1) | 0,45 | 0,17 | 0,019 | 1,9 | 3,0 | 5,7 | 4,3 |
Tableau 9.5. Viscosités et permittivités de quelques solvants
| Solvant | Viscositéa h | Permittivité er | Solvant | Viscositéa h | Permittivité er |
| hexane | 0,326 | 1,89 | furfural | 1,49 | 41,9 |
| méthanol | 0,597 | 32,63 | propanol | 2,26 | 20,1 |
| tétrachlorure de carbone | 0,969 | 2,23 | butanol | 2,95 | 17,1 |
| eau | 1,00 | 78,54 | formamide | 3,3 | 109 |
| cyclohexane | 1,02 | 2,01 | aniline | 4,40 | 6,89 |
| éthanol | 1,20 | 24,3 | glycol | 19,9 | 37,7 |
| glycérol | 1490 | 42,5 | |||
| a : 103 kg·m-1·s-1 (voir Handbook of Chemistry and Physics, 62e Édition, 1981-82). | |||||
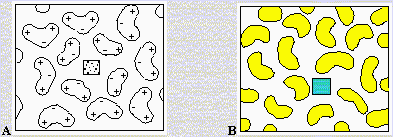
Le solvant joue maintenant un rôle crucial. En effet, les charges portées par l’un ou l’autre, ou les deux, réactifs développent un champ central qui est perturbé par les molécules de solvant selon la permittivité de ce dernier. Dans le cas d’un liquide de permittivité élevée, le champ de l’ion réactif ne se fera sentir qu’à courtes distances (environ 0,7 nm dans l’eau) alors que dans un solvant comme l’hexane il se fera sentir jusqu’à 30 nm. Ainsi les réactions entre ions de charges opposées seront-elles plus rapides dans un solvant de faible permittivité (Fig. 9.2 A). Cette figure permet de visualiser l’écran constitué par les molécules de solvant autour de l’ion. Dans le cas de Fig. réaction entre deux ions chargés de même signe, les réactions dans un solvant de faible permittivité seront plus lentes puisque les forces de répulsion coulombienne se font sentir à plus grande distance (Fig. 9.2 B).
|
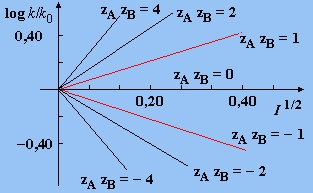
La loi de DEBYE-HÜCKEL a conduit à exprimer la variation de la constante de vitesse par la relation (lorsque la réaction a lieu dans l’eau à 25 °C : A = 1,02) :
| 9.4 |
log k = log k0 + |
|
Dans cette équation I est la force ionique de la solution. Dans le cas des solutions aqueuses diluées, lorsque la force ionique est très faible, l’expression précédente se réduit à :
log k = log k0 + 1,02 zA zB (I)1/2
La force ionique de la solution est donnée par la relation :
| 9.5 |
I = 1/2 S Ci zi2 |
Le terme Ci représente la concentration de l'ion i portant un nombre z de charges électriques négatives ou positives.
|
|
Figure 9.3. Variation de la vitesse de réaction en
solution
en fonction de la force ionique.
Soit l’équilibre
![]()
Et soient k1 et k-1 les constantes de vitesse des réactions directe et inverse. La constante d’équilibre peut s’écrire ainsi :
Ln K = - DG° / RT
On sait en outre (voir le cours de thermodynamique) que :
![]()
DV° = SV°(produits) - SV°(réactifs)
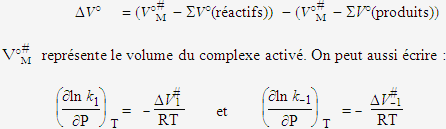
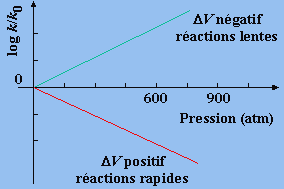
Il apparaît que la constante de vitesse de la réaction croît avec l’augmentation de pression si le volume du complexe activé est plus petit que le volume total des réactifs, et inversement (Fig. 4). De plus la variation de ln k en fonction de la pression doit être une droite dont la pente est égale à
- D V# / RT
|
|
Figure 9.4. Variation de la vitesse de réaction
en solution en fonction de la pression.
On dispose ainsi d’une méthode simple pour mesurer la variation entre le volume des réactifs et celui du complexe activé (Fig. 9.5). Le tableau qui suit montre quelques valeurs numériques.
|
|
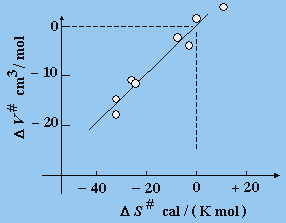
Fig. 9.5. Variation de volume et d’entropie d’activation dans une réaction en solution.
Tableau 9.6. Variation du volume d'activation
| Réaction chimique | Solvant | DV# | ||
| N(C2H5)4+ + OH- | ® | CH3OC2H5 + N(C2H5)3 | CH3OH | + 20 |
| C6H5COOOOCC6H5 | ® | 2 C6H5COO | CCl4 | + 11 |
| Co(NH3)5Br2+ + OH- | ® | Co(NH3)5OH2+ + Br- | H2O | + 8,5 |
| Sucrose + H2O | ® | glucose + fructose | H2O (H+) | + 2,5 |
| C2H5O- + C2H5I | ® | C2H5OC2H5 + I- | C2H5OH | - 4,1 |
| CH3COOCH3 + H2O | ® | CH3COOH + CH3OH | H2O (H+) | - 8,7 |
| CH3CONH2 + H2O | ® | CH3COOH + NH3 | H2O (OH-) | - 14,2 |
| 2 cyclopentadiène | ® | C10H12 | n-C4H9Cl | - 22 |
Note : DV# en cm3·mol-1.
Incidemment, il faut remarquer que le remplacement de deux moles de cyclopentadiène par une mole de dicyclopentadiène représente une diminution de volume égale 22 cm3, soit à une sphère de 0,3 nm par molécule. Ce qui est dans l’ordre de grandeur réelle, soit celle d’une molécule de grosseur voisine à celle du cyclopentadiène dont le rayon moyen est de 0,4 nm :
4/3 p r3 N = 22 cm3 et r = 0,3 nm
Cette variation de volume est accompagnée d’une variation d’entropie (Fig. 9.5).
On peut distinguer trois cas :
a. La constante de vitesse augmente lorsqu’on accroît la pressionDV# = V# - VR < 0
Les grandeurs VR et V# sont respectivement les volumes des réactifs et du complexe activé. Le volume diminue lors de la formation du complexe activé.
Cela est interprété comme une augmentation de la charge globale lors de la formation du complexe activé. Ce phénomène de diminution du volume par augmentation de la charge s’appelle électrostriction. Si un ion est dissout dans un solvant polaire, le champ électrostatique des ions cause une orientation des dipôles du solvant. L’orientation des molécules du solvant permet un arrangement mieux ordonné de ce dernier et une minimisation de l’espace occupé.
Ce phénomène s’observe pour les substitutions nucléophiles unimoléculaires SN1. Dans une solution eau - éthanol, la réaction suivante de premier ordre est un processus SN1 :
R - X ® R+ + X- (étape lente)
NaOH + (CH3) 3CCl ® (CH3) 3COH + NaOH
Le complexe activé de l’étape lente implique la séparation de charge de RX.
b. La pression a peu d’effet sur la constante de vitesse
DV# = V# - VR = 0
Cela se produit lorsque l’état de transition n’implique pas de modification de charges par rapport aux réactifs. Ce phénomène s’observe pour les substitutions nucléophiles bimoléculaires SN2 :
NaOH + CH3CH2Cl ® CH3CH2OH + NaCl
La réaction est une seule étape élémentaire impliquant la formation du complexe activé formé des deux molécules de réactifs.
c. La constante de vitesse diminue avec l’augmentation de la pression
DV# = V# - VR > 0
Cela se produit lorsque les réactifs ont des charges opposées et que le complexe activé formé possède une charge inférieure à celle de la somme en valeurs absolues des réactifs.
Co(NH3) 5Br2+ + OH- ® Co(NH3) 5OH2+ + Br-
Si l’électrostriction est importante (cas a), l’ordre des molécules de l’état transitoire sera plus grand et la variation d’entropie est négative
DS# = S# - SR < 0
k = ( R T ) /( N h) e+D S#/R e-D H#/RT
Cette expression explique pourquoi la constante de vitesse diminue avec l’augmentation d’électrostriction. L’effet de la pression est beaucoup plus faible que l’effet de la température. Même pour des valeurs de DV# importantes (± 0 cm3·mol-1), cela demande des pressions énormes, de l’ordre de 430 atmosphères pour doubler la constante de vitesse.
Physiquement, on peut expliquer que les réactions qui se font avec une diminution de volume (entre les réactifs et le complexe activé) ont une vitesse qui augmente avec l’augmentation de pression. Il y a diminution de l’entropie et les réactions sont normalement lentes. Inversement, lorsqu’il y a augmentation de volume, l’augmentation de pression joue contre la vitesse de réaction et l’entropie augmente au cours de la réaction. Les vitesses de réaction sont dans ces cas normalement rapides. Le tableau suivant résume ces résultats.
Tableau 9.7. Classification des
réactions selon les variations
des volumes d’activation
| DV# | DS# | exemples de réactions | classification |
| >0 » 0 <0 |
>0 » 0 <0 |
entre ions de même signe entre molécules neutres entre ions de signe opposé |
réactions lentes réactions normales réactions rapides |
La
sonochimie est la chimie assistée ou provoquée par l’effet des ultrasons.
En effet, un liquide soumis à un passage d’ondes sonores développe
des microcavités qui augmentent et diminuent alternativement de volume, selon
la fréquence du champ appliqué. Ces
bulles microscopiques de l’ordre du micromètre apparaissent en quelques
microsecondes avant de s’effondrer sur elles-mêmes.
Comme les échanges de chaleur avec le milieu sont relativement lents, il
peut s’y développer des températures pouvant dépasser 5 000 K et des
pressions de l’ordre de 1 000 bars voire plus !
Il s’agit de conditions expérimentales relativement rares ou
difficiles (à coût élevé) à provoquer de manière continue dans un réacteur.
Ces bulles microscopiques peuvent donc être le siège de réactions bien
particulières. La dimension, le temps de vie et le comportement de ces cavités
dépendent de la fréquence, de l’intensité (la pression acoustique), du
solvant, et de paramètres externes comme la température et la pression. On
observe dans certains systèmes une luminescence appelée sonoluminescence.
On
divise arbitrairement le spectre sonique qui va de 20 kHz à 10 MHz en trois régions :
1-
la région de basse fréquence ou de haute énergie : 20 à 100 kHz;
2-
la région de haute fréquence et d’énergie moyenne : 100 kHz à
1 MHz; et
3-
la région de haute fréquence et de basse énergie : 1 à 10 MHz..
Les
deux premières régions sont celles de la sonochimie tandis que l’usage de la
troisième est celle dans le domaine médical et celui du diagnostique.
Exemple
de réactions affectées par les ultrasons : l’époxydation des liaisons
carbone-carbone insaturées. La
vitesse de la réaction suivante :
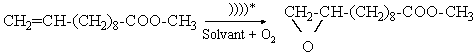
* : symbolisme utilisé pour indiquer l’usage d’ultrasons.
En quatre fois moins de temps (25 min au lieu de 2 heures), le rendement de la réaction est multipliée par 2 (94 au lieu de 48 %).
Dans une solution eau-éthanol, la constante de vitesse de la réaction d’hydrolyse en phase homogène du chlorure de tertio-butyle augmente sensiblement (Tableau 9.8) en présence d’ultrasons (20 kHz) :
(CH3)
3C-Cl
+ H2O ®
[(CH3) 3C+ + Cl-]
®
(CH3) 3C-OH + HCl
Tableau 9.8. Hydrolyse du chlorure de tertio-butyle
| Constante de vitesse relative | 10 °C | 25 °C |
| ksilence | 0,86 | 6,8 |
| kultrason | 17,2 | 11,4 |
| Rapport | 20,0 | 1,7 |
| Milieu : éthanol-eau (40-60, poids/poids). | ||
Tiré
de : Mason,
T.
J., Elements of organic chemistry of interest to sonochemists, in
« Sonochemistry
and Sonoluminescence », L. A.
Crum, T. J.Mason, J. L. Reisse et K. S. Suslick, éditeurs, Kluwer Academic
Publishers, London, 1999.
Dans ce cas, l’application d’ultrasons augment la vitesse de réaction et contrairement à ce que l’on sait de la cinétique, la vitesse de réaction est plus élevée à 10 °C qu’à 25 °C. Ces données sont révélatrices de comportements surprenants qui indiquent que les connaissances théoriques et pratiques de l’utilisation des ultrasons sont encore loin d'être bien comprises.
8. Conclusions
|
9. Exercices
| 1 - | L’oxydation des iodures par l’eau oxygénée est le résultat de deux réactions telles que la vitesse de réaction est donnée par la relation : |
| = k1 [H2O2] [I -] + k2 [H2O2] [I -] [H +] |
La force ionique à 25 °C influence les constantes de vitesse de la manière suivante [Bell, F. et al., J. Phys. Chem., 55, 874 (1951)] :
| Force ionique | 0,00 | 0,0207 | 0,0525 | 0,0925 | 0,1575 | 0,2025 |
| k1, mol-1 · s-1 | 0,658 | 0,663 | 0,670 | 0,679 | 0,694 | 0,705 |
| k2, mol-1 · s-1 | 19,0 | 15,0 | 12,2 | 11,3 | 9,7 | 9,2 |
Évaluer graphiquement la validité de l’équation 9.3 dans chacun des deux cas. Dites ce que vous en pensez.
| 2 - | Pour la réaction NH2SO2OH + H2O ® NH4HSO4 en solution à 80,5 °C, la constante de vitesse a été mesurée à différentes forces ioniques. À partir des résultats qui suivent, déterminez la valence des ions qui réagissent. À cette température, le facteur A est égal à 0,574. |
| Force ionique | 0,005 06 | 0,011 21 | 0,015 85 | 0,022 94 |
| k1, mol-1 · h-1 | 1,07 | 1,02 | 0,976 | 0,886 |
| 3 - | La constante de vitesse, k (× 105 M-1·s-1), d’une réaction dépend de la pression (T = 52,5 °C) : |
| Pression (atm) | 1 | 1 500 | 2 300 | 2 600 | 2 875 | 4 925 |
| Solvant : méthanol | 3,18 | 12 | 27,7 | 57,6 | ||
| Solvant : nitrophénol | 2,24 | 6,15 | 9,51 | 11,3 |
Estimez la variation DV# de la réaction à la pression normale.
4- Une réaction donne les résultats suivants :
| T (K) | 173 | 183 | 193 | 203 |
| k (l· mol-1·s-1 | 34 | 150 | 500 | 1 800 |
Calculez le facteur pré exponentiel et l’énergie d’activation de cette réaction.
|
5 -
|
La réaction [Co(NH3) 5NO2] 2+ + OH- ® [Co(NH3) 5 OH] 2+ + NO2- a été étudiée en solution et on a trouvé que sa constante de vitesse varie en fonction de la force ionique de la solution. À l’aide des données fournies au tableau, trouver graphiquement k0, la constante de vitesse correspondant à une force ionique nulle.
Montrer ensuite que le comportement de cette réaction est conforme à ce que prédit la théorie. Les unités de k sont en L mol-1 s-1. |
|
6 -
|
L’étude de la réaction [Co(NH3)5Br]2+ + OH- ® [Co(NH3)5OH]2+ + Br- en solution aqueuse, à 15 °C, et à des concentrations initiales de 0,5 10-3 M pour l’ion [Co(NH3)5Br]2+ (présent sous forme de bromure) et de 0,705 10-3 M pour l’ion OH- (présent sous forme de NaOH), a permis de trouver la constante de vitesse de cette réaction : k = 91 L mol-1 min-1 À partir de ce résultat, déterminer la constante de vitesse à dilution infinie. Calculer ensuite les constantes de vitesse de cette réaction pour les différentes concentrations des deux ions quand la réaction est faite en présence de quantités différentes de NaCl (voir tableau). Comparer ces résultats avec ceux trouvés expérimentalement.
|
|
7 -
|
Lors de l’étude de la réaction 2 [CoBr(NH3)5]2+ + Hg2+ + 2 H2O ® 2 [Co(NH3)5H2O]3+ + HgBr2 les résultats suivants ont été obtenus :
À partir de ces résultats déterminer graphiquement k pour une solution contenant 4 10-4 mol·L-1 de [CoBr(NH3)5]2+, 2,78 10-4 mol·L-1 de Hg2+ et 1,98 10-2 mol·L-1 de HNO3. Comparer ce résultat à la valeur obtenue expérimentalement (k = 250 L·mol-1·min-1). Note : l’ion [CoBr(NH3)5]2+ est présent sous forme de bromure et l’ion Hg2+ est présent sous forme de nitrate.
|
|
8 -
|
La réaction S2O82- + 2 I- ® 2 SO42- + I2 a été faite à partir d’une concentration initiale de persulfate de sodium égale à 1,5 ´ 10-4 mol L-1. Les résultats expérimentaux ainsi obtenus sont les suivants :
1- Estimer ZAZB pour cette réaction et calculer la constante de vitesse quand les coefficients d’activité sont égaux à l’unité. 2- Est-ce que la force ionique de la solution varie au cours de la réaction ? |
| 9
-
|
La vitesse de saponification de l’acétate de méthyle à 25 °C est étudiée en partant d'une solution 0,01 molaire en ester et en base. On titre le mélange à différents temps avec une solution standard d’acide. Les résultats sont les suivants : |
| Temps (min) | 3 | 5 | 7 | 10 | 15 | 21 | 25 |
| Concentration observée* | 0,0074 | 0,00634 | 0,00550 | 0,0046 | 0,00363 | 0,00288 | 0,00254 |
*: On ne donne pas dans quelles unités sont exprimées ces concentrations.
|
|
Montrer par une méthode graphique que la réaction est d’ordre 2 et Déterminez la constante de vitesse de cette réaction. |
Pour en savoir un peu plus :
Weston, R. E., Jr. et H. A. Schwarz, Chemical Kinetics, Prentice-Hall, inc., New Jersey, 1972.
Un document bien fait en "espagnol" : Rodriguez Velasco, J., F. Sanchez Burgos et M. Dominguez Perez, Lecciones de cinética química, Université de Séville, Sevilla (Espagne), 1975.
Une revue de réactions observées en chimie organique en présence d’ultrasons : "Synthetic organic sonochemistry", Jean-Louis Luche, éditeur, Plenum Press, New York, 1998, 431 p.
Sur le NET :
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes industries, les Fondations, ...
Dernière mise à jour : 2021-07-01.