|
|
CHAPITRE 7
LA CHIMIE DES FLAMMES
|
Cas industriellement importants, et aussi tout autant dans la vie domestique, les flammes sont omniprésentes dans la vie courante. Questions :
|
Objectifs spécifiques
- Comprendre les mécanismes de réactions chimiques intervenant dans les systèmes d’oxydation dégageant de grandes quantités d’énergie.
- Appliquer les principes et les lois de la cinétique aux flammes ainsi qu’aux anomalies dont elles peuvent être l’objet.
- Comprendre les phénomènes de production de suie, de luminescence, d’ions dans les flammes.
La flamme est un milieu très particulier dans laquelle une multitude de réactions chimiques ont lieu. Caractérisés par un dégagement de chaleur intense, ces milieux sont d’un emploi industriel, commercial, domestique on ne peut plus universel. Ces réactions méritent donc une attention particulière d’autant que, crise de l’énergie aidant, elles sont soumises à de nombreuses études afin, dans un premier temps, de bien les connaître, mais aussi et surtout d’en améliorer les qualités. On verra donc non seulement les réactions d’oxydation, mais également les processus d’émission de lumière, les processus ioniques,...
2. Le mécanisme réactionnel
Le mécanisme réactionnel de base de l’oxydation des combustibles hydrocarbonés est similaire à celui de l’oxydation aéronomique de ces mêmes molécules avec les particularités d’une flamme à température élevée. Compte tenu de la spontanéité et de la vitesse de ces oxydations, on imagine très bien l’implication d’un mécanisme en chaîne, probablement ramifiante, puisque l’on assiste dans des conditions mal contrôlées à des explosions. Soit donc le mécanisme général suivant :
| [1] | RH + O2 | ® R• + HO2• | DH = 190-210 kJ/mol |
| [2] | R• + O2 | ® RO2• | Ea = 0 kJ/mol |
| [3] | R• + O2 | ® alcène + HO2· | |
| [4] | RO2• + RH | ® ROOH + R• | |
| [5] | RO2• | ® R'CHO + R"O• | |
| [6] | HO2• + RH | ® H2O2 + R• | |
| [7] | ROOH | ® RO• + •OH | |
| [8] | R'CHO + O2 | ® R'CO• + HO2• | DH = 134 -138 kJ/mol |
| [9] | RH + •OH | ® H2O + •R | |
| [10] | R'CO• | ® R' + CO | |
| [11] | R'CO• + O2 | ® R'C(=O)-OO• | |
| [12] | RO2• | ® parois ... etc. | Ea = 0 kJ/mol |
La réaction [1] constitue l’étape d’amorçage de la réaction d’oxydation de l’hydrocarbure : c’est une réaction endothermique. Les réactions [2] à [6] et [9] à [11] constituent des étapes de propagation linéaire et les réactions [7] et [8] des étapes de propagation ramifiante. En phase gazeuse, la réaction [5] est plus rapide que la réaction précédente [4]. Dans le cas du méthane, les réactions [3] et [4] sont évidemment impossibles. Le mécanisme, on le voit, dépend de la nature de chaque combustible. Il faut ajouter que la nature du comburant a lui aussi ses conséquences. Comme autres paramètres, on peut ajouter la pression totale, les concentrations relatives du combustible et du comburant, la forme du réacteur ou du brûleur, etc.
On identifie assez facilement les différentes étapes du mécanisme en chaîne. Dépendant de la température, de la pression, de la nature des parois, on obtient comme dans le cas du mélange hydrogène - oxygène des zones de stabilité relative et des zones d’instabilité (oxydation brutale) - voir Chapitre 4.7. Ainsi dans le cas du mélange propane - oxygène (1 : 1), on peut observer ces zones dans les conditions décrites à la figure 7.1. À la pression atmosphérique, le mélange est stable pourvu que la température soit inférieure à 200 °C. Au-dessus de 250 °C, le mélange explose naturellement.
| [a] | CH4 + O2 | ® CH3• + HO2• | DH = 190-210 kJ/mol |
| [b] | CH3• + O2 | ® CH3O2• | Ea = 0 kJ/mol |
| [c] | CH4 + •OH | ® H2O + CH3• | |
| [d] | CH3O2• | ® HCHO + HO• | |
| [e] | HO• + HCHO | ® H2O + HCO• | |
| [f] | HCHO + O2 | ® CO + HO2• | |
| [g] | CH4 + HO2· | ® H2O2 + CH3• | |
| [h] | HCHO + HO2• | ® H2O2 + HCO• | |
| [i] | •OH | ® parois | |
| [j] | HCHO | ® parois |
3. Réactions explosives
La réaction entre l’hydrogène et l’oxygène possède des caractéristiques différentes de celles des réactions qui ont été étudiées au chapitre 4. En effet, dans le cas de la réaction entre H2 et O2, tout comme dans le système méthane-oxygène, les conditions pour lesquelles le mélange devient explosif sont différentes de celles décrites dans le cas des réactions en chaîne linéaire ou ramifiante. Par conséquent, on doit chercher, pour cette auto oxydation, un mécanisme spécial à partir duquel la réaction se développe. Cependant, avant de décrire les caractéristiques de cette réaction, il faut préciser la notion d’explosion.
Une explosion thermique se produit quand la principale cause de l’auto accélération de la réaction est l’auto échauffement du mélange réactionnel, causé par une réaction exothermique. En général, dans le cas d’une réaction exothermique, la température d’un élément de volume du mélange réactionnel se stabilisera quand la vitesse de production de chaleur par la réaction sera égale à la vitesse de perte de chaleur par conduction, convection ou radiation. La vitesse de production de chaleur, QR, dans un volume de gaz, V, réagissant à une vitesse par unité de volume, R, est donnée par :
| 7.1 |
QR = - V R DH |
Dans cette équation DH est la quantité de chaleur dégagée par mole de réaction. Si on pose :
R = k Cn (réaction d’ordre n)
On peut écrire en développant la valeur de la constante de réaction k :
| 7.2 |
QR = - V Cn A e-Ea/RT DH |
A est le facteur pré-exponentiel de la réaction
Ea est l'énergie d’activation, et
C est la concentration du réactif
La vitesse de perte de chaleur QC dépend de façon complexe des paramètres physiques du système. Cependant, pour de faibles différences entre la température du gaz, T, et celle de son environnement, TS, la loi empirique de Newton peut être appliquée :
| 7.3 |
QC = S h (T - TS) |
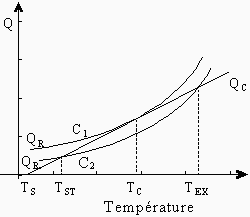
La grandeur S est la surface du réacteur et h est le coefficient de transfert de chaleur par unité de surface. À la figure 7.1, on peut voir comment QR et QC varient selon la température du gaz.
|
|
Figure 7.1. Variation de QR et QC en fonction de la température.
D’après la courbe C2 de la figure 7.1, on peut déduire que si un mélange de concentration C2 est introduit dans le réacteur à température TS, QR sera plus grand que QC. Il y aura alors élévation de la température du gaz jusqu’à ce qu’il atteigne la température TST, c’est-à-dire la température à laquelle les courbes QR et QC se croisent. À cette température, le système est stable: si un petit changement des conditions auxquelles se trouve le système survenait et que sa température montait légèrement, le système retournerait à la température TST car QC est alors plus grand que QR. Inversement, si la température tombait légèrement, la température du système tendrait à remonter à TST (car QR > QC). Par contre, si la température du système était élevée à une valeur supérieure à TEX la température d’explosion, le système ne serait plus stable. À ce moment QR est supérieur à QC et la température du mélange croît sans limite (ou en pratique jusqu’au moment où la vitesse de la réaction est suffisamment ralentie par la diminution de la concentration des réactifs).
La courbe C1 présente une situation particulière: les courbes QC et QR se touchent à la température TC où TST et TEX coïncident. Cette température critique, TC, est la plus haute à laquelle une réaction stable peut se produire, et cette concentration critique, C1, est donc la plus basse à laquelle une explosion thermique spontanée peut se produire. La courbe C1 est ainsi appelée une limite d’explosion, car elle sépare la zone où la réaction est stable de la zone où la réaction est explosive. Le calcul de TC peut être fait ainsi :
QR = QC
alors,
d QR / dT = d QC / dT
ce qui donne :
| 7.4 |
TC - TS = RTC2 / Ea |
Le calcul de CC se fait directement à partir de l’équation 7.2, c’est-à-dire :
| 7.5 |
S h (TC - TS) = - V CCn A e-Ea/RT DH |
On peut, de cette façon, calculer les conditions critiques d’une explosion thermique si on connaît les dimensions du réacteur, le coefficient de transfert de chaleur et les paramètres cinétiques et thermodynamiques de la réaction. On peut ainsi prédire sous quelles conditions une réaction en chaîne linéaire peut devenir explosive, c’est-à-dire, à quelle température il y aura auto-accélération de la réaction consécutive à son auto-échauffement.
L’étude de la réaction entre l’hydrogène et l’oxygène montre des caractéristiques qui ne peuvent s’interpréter en termes de chaînes linéaires. Les données expérimentales de base sont les suivantes :
À basse température, la réaction est fortement hétérogène (elle se produit à la surface de tout solide présent plutôt que dans la phase gazeuse) et aux températures supérieures à 400 °C, elle peut être explosive en phase gazeuse. Au-dessus de 600 °C, elle est presque toujours explosive.
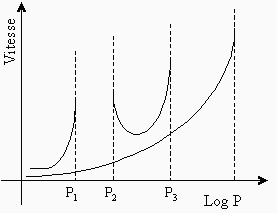
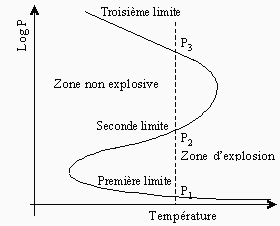
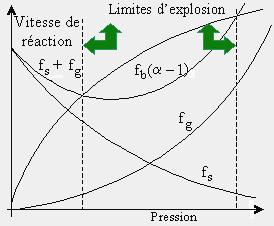
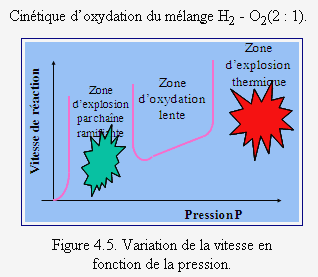
À 550 °C, la vitesse de réaction d’un mélange stœchiométrique de H2 et O2 varie de façon très particulière en fonction de la pression du mélange (voir Fig. 7.2). On remarque sur cette figure que l’allure de la courbe supérieure, particulièrement aux points P1, P2 et P3, est très différente de celle de la courbe inférieure qui correspond à une réaction en chaîne linéaire. L’étude de cette figure montre qu’à très basse pression, la réaction évolue très lentement (la réaction est stable). Si on augmente ensuite la pression, la vitesse de la réaction s’accélère puis devient explosive (au point P1). Cette pression représente alors sa première limite d’explosion (ou limite inférieure d’explosion). Si on continue à augmenter la pression, la réaction demeure explosive, puis, à une pression donnée (au point P2), la vitesse de la réaction commence à diminuer très rapidement. La réaction passe à ce point sa seconde limite d’explosion (ou limite supérieure d’explosion). Ensuite, à mesure que la pression du mélange est augmentée, la vitesse de la réaction diminue pour atteindre, un peu plus loin, une région de pressions où elle recommence à croître. Au point P3, la réaction est redevenue explosive. Elle a atteint sa troisième limite d’explosion. Pour chaque température, on obtient ainsi trois limites d’explosion, et les variations de ces limites, avec la température, peuvent être représentées par un diagramme qui dépend en partie du rapport surface/volume et de la nature du réacteur (voir Fig. 7.3). La partie gauche du diagramme d’explosion représente la zone non explosive de la réaction, alors que la partie droite est la région où se produisent les explosions (voir aussi Fig. 4.5).
|
|
Figure 7.2. Limites d’explosion pour une réaction en chaîne ramifiée.
La limite inférieure d’explosion s’abaisse lentement quand la température du mélange est accrue ou qu’un gaz inerte est ajouté au mélange. Elle est très sensible à la nature de la surface du réacteur et est inversement proportionnelle au diamètre du réacteur. Par contre, la limite supérieure d’explosion s’élève rapidement avec l’augmentation de la température selon une énergie d’activation apparente de 20 à 25 kcal mol-1. La seconde limite peut être abaissée par l’addition d’un gaz inerte et particulièrement d’eau. Elle n’est presque pas affectée par la nature et les dimensions du réacteur.
La troisième limite est plus difficile à définir expérimentalement car elle correspond à une région où la vitesse est très rapide dans la zone non explosive. De plus, cette limite est située près de la limite thermique d’explosion, où beaucoup d’auto-échauffement se produit. Cette limite décroît très rapidement avec la température et elle est abaissée par l’addition de gaz inerte et par l’augmentation du diamètre du réacteur.
Figure 7.3. Diagramme d’explosion pour un mélange stœchiométrique d'hydrogène et d'oxygène.
Les caractéristiques très particulières de la région située entre P1 et P2 énumérées ci-dessus pour la réaction entre H2 et O2 ne peuvent être attribuées à une réaction qui se fait suivant un mécanisme en chaîne linéaire. En effet, la propagation de la chaîne ne faisant pas intervenir la pression, on ne devrait pas avoir trois limites d’explosion, mais une seule et tributaire uniquement de la température. Il faut donc admettre que cette réaction s’effectue par un mécanisme différent, et que les réactions explosives qu’elle manifeste sont d’un autre type que celles qui se produisent lors des explosions thermiques. On doit alors conclure qu’elle se fait par un type de réaction en chaîne où un centre propagateur, au lieu de se renouveler simplement, peut en créer plusieurs autres et participer à plusieurs réactions élémentaires. Un tel processus constitue ce qu’on appelle une réaction en chaîne ramifiée. Ce type de mécanisme donne lieu à des explosions dites isothermes.
Dans les explosions isothermes, la principale cause de l’auto-accélération est la multiplication des centres propagateurs due aux processus de ramification de la chaîne. Le qualificatif isotherme ne signifie pas que le processus de l’explosion est vraiment isotherme, mais tout simplement que l’explosion aurait aussi lieu si on parvenait, par un quelconque moyen, à garder constante la température des réactifs. La cause fondamentale de l’explosion est donc chimique et non thermique en nature. Par conséquent, les caractéristiques des explosions isothermes sont très différentes de celles des explosions thermiques.
Une illustration très élémentaire des principes qui régissent le développement des réactions en chaînes ramifiées peut être faite à partir du mécanisme suivant :
| [1] | ? ® R• | Initiation (i) |
| [2] | R• ® a R• | Ramification (b) |
| [3] | R• ® P + R• | Formation de produits (p) |
| [4] | R• ® ? | Rupture sur les parois (s) |
| [5] | R• ® ? | Rupture en phase gazeuse (g) |
La première réaction représente la production initiale de radicaux. La réaction 2 est l’étape de ramification où le radical R induit, par un processus élémentaire, la production de plus d’un radical. Il en résulte un accroissement très rapide du nombre de radicaux et alors, les conditions de l’état stationnaire ne tiennent plus. La réaction se produit alors à une très grande vitesse et une explosion se produit. Les produits sont formés par la réaction 3 qui produit aussi, dans beaucoup de cas, un autre radical.
On peut alors établir les équations qui représentent la vitesse des différentes étapes du mécanisme. Afin de simplifier le problème, considérons que toutes les réactions sont du premier ordre. Les vitesses des réactions peuvent être écrites ainsi:
| (1) | Vi = d[R•]/dt |
| (2) | Vb = d[R•]/dt = fb(a-1)[R•] |
| (3) | Vp = d[P]/dt = fp[R•] |
| (4) | Vs = - d[R•]/dt = fs[R•] |
| (5) | Vg = - d[R•]/dt = fg[R•] |
Les coefficients fb, fg, fp et fs peuvent être fonction des concentrations des réactifs, des produits ou d’autres substances présentes. L’équation stationnaire pour R est :
| 7.6 |
Vi + fb(a-1)[R•] - (fs+fg)[R•] = 0 |
D’où :
![]()
La vitesse globale de la réaction définie comme
|
est donc |
|
Comme a (le coefficient de ramification) est toujours supérieur à l’unité, le dernier terme du dénominateur de l’équation précédente est positif, mais il peut être nul sous certaines conditions. À ces conditions, le numérateur devient infiniment grand et donc, la vitesse tend vers une valeur infinie. Elle devient donc très grande en pratique. Cependant, à ce moment, les conditions requises pour un état stationnaire ne sont plus remplies. Néanmoins, la condition
fb(a-1) = fs + fg
peut être utilisée pour déterminer l’approche de la limite d’explosion. Les grandeurs relatives de fs, fg et fb(a-1) dépendent des concentrations des différentes espèces présentes dans le mélange réactionnel. On peut voir, à la figure 7.4, comment varient les coefficients fs et fg en fonction de la pression des réactifs, et sous quelles conditions se trouvent les limites d’explosion.
|
|
Figure 7.4. Détermination des limites d’explosions dans les cas de mécanismes simples à chaîne ramifiée.
Le mécanisme détaillé de la réaction entre H2 et O2 est très complexe car il fait intervenir un très grand nombre de réactions élémentaires. Cependant, on peut expliquer les grandes caractéristiques de la réaction à partir du mécanisme simplifié de HINSHELWOOD :
| [1] | H2 | ® 2 H• | k1 |
| [2] | H• + O2 | ® •OH + O• | k2 |
| [3] | O• + H2 | ® •OH + H• | k3 |
| [4] | H• + O2 + M | ® HO2• + M | k4 |
| [5] | HO2• | ® destruction sur les parois | k5 |
| [6] | HO2• + H2 | ® H2O + •OH | k6 |
| [7] | •OH + H2 | ® H2O + H• | k7 |
| [8] | •H | ® destruction sur les parois | k8 |
| [9] | •OH | ® destruction sur les parois | k9 |
L’initiation de la chaîne se fait par la réaction [1] et la réaction se ramifie ensuite par les étapes [2] et [3]. On a ainsi une ramification linéaire dont le coefficient de ramification, a, est 3 (un atome d’hydrogène, par suite des réactions [2] et [3], produit 2 radicaux OH• et un autre atome d’hydrogène). Ce processus donne ainsi lieu à une telle multiplication des actes élémentaires que ceux-ci sont responsables de l’allure explosive prise par la réaction dans toute la partie droite de la courbe de la figure 7.3.
Au voisinage de la seconde limite d’explosion, il y a rupture de la chaîne par les réactions [4] et [5]. L’existence de la première et de la seconde limite d’explosion s’explique donc par le fait que la rupture de chaîne peut se faire soit sur les parois du réacteur ou soit dans la phase gazeuse. Le processus de rupture de chaîne sur les parois est alors dominant: les réactions [8] et [9] sont donc importantes à ce moment tandis que les réactions [4] à [6] sont négligeables; les réactions de rupture sont plus nombreuses que les réactions de propagation et la réaction ne peut être explosive.
Les équations stationnaires aux basses pressions sont les suivantes :
[A] Vi - k2[H•][O2] + k3[O•][H2] + k7[•OH][H2] - k8[H•] = 0
[B] k2[H•][O2] - k3[O•][H2] = 0
[C]
k2[H•][O2]
+ k3[O•][H2] -
k7[•OH][H2]
-
k9[•OH]
= 0
[D] Vi + k7[•OH][H2] - k8[H•] = 0
[E] 2 k2[H•][O2] - k7[•OH][H2] - k9[•OH] = 0
Des équations [D] et [E] on peut tirer :
![]()
Sous ces conditions, la vitesse de réaction (vitesse de formation de H2O) s’écrit donc :
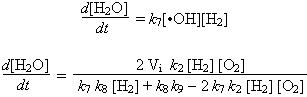
Dans le dénominateur de cette fraction, la somme k7 k8 [H2] + k8 k9 exprime les processus de rupture sur les parois.
Si la pression initiale des réactifs est accrue, la probabilité qu’un radical atteigne les parois diminue (fs diminue). Ainsi, au fur et à mesure qu’on augmente la pression, le nombre moyen de collisions multiplicatives de chaque porteur de chaîne augmente, alors que la vitesse de diffusion des radicaux vers les parois diminue d’autant. Le dénominateur de la dernière expression devient de plus en plus petit (il tend vers zéro) et la vitesse s’accélère jusqu’à devenir explosive: la probabilité de multiplication des chaînes excède la probabilité de rupture des chaînes. Si on continue d’accroître la pression des réactifs, les réactions [8] et [9] deviennent négligeables: peu de radicaux H• et •OH atteignent la paroi. La réaction trimoléculaire [4] prend de plus en plus d’importance, et les radicaux HO2• alors formés sont en grande partie détruits sur les parois (les radicaux HO2• étant relativement stables sous ces conditions, leur probabilité d’atteindre les parois est suffisamment grande à ces pressions pour justifier ce mécanisme). Comme trop peu de radicaux HO2• réagissent alors par la réaction [6], la réaction [4] induit donc une rupture de chaîne et la réaction cesse d’être explosive. On a atteint la seconde limite d’explosion. Comme l’expérience montre que la valeur de cette seconde limite dépend pour une large part des dimensions du récipient, on confirme ainsi l’intervention d’une réaction telle que la réaction [1].
Si la pression des réactifs est encore accrue, la stabilité des radicaux HO2• diminue: leur vitesse de diffusion vers les parois diminue et leur probabilité de réaction par la réaction [6] augmente. À mesure que la pression augmente, la réaction [6] réactive de plus en plus la chaîne alors que les ruptures de chaîne sur les parois sont de moins en moins nombreuses. La réaction redevient explosive. De plus, aux pressions les plus hautes, il y a une contribution thermique à l’explosion.
Le même type de mécanisme a été appliqué pour expliquer la combustion du monoxyde de carbone et des sulfures d’hydrogène et de phosphore. Cependant, la combustion des hydrocarbures se fait par un mécanisme différent de celui indiqué plus haut.
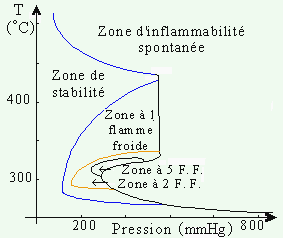
Dans le cas du mélange propane - oxygène (1 : 1), on peut observer ces zones dans les conditions décrites à la figure 7.5. À la pression atmosphérique, le mélange est stable pourvu que la température soit inférieure à 200 °C. Au-dessus de 250 °C, le mélange explose naturellement.
|
|
Figure 7.5. Zones de stabilité et d’instabilité du mélange C3H8 -
O2 (1 : 1).
(F. F. : flammes froides) Note : les zones à 3 et 4 F.F. ont été ignorées par souci de
clarté.
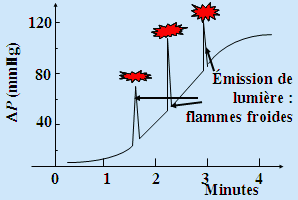
La figure précédente montre aussi une région perturbée. En effet, dans la péninsule de stabilité, on observe l’émission spontanée et brutale de lumière : ce phénomène est appelé l’émission de flamme froide. Cette émission peut se produire 1, 2, 3, n fois dépendant des conditions expérimentales, du couple comburant - combustible,... Par souci de clarté, on a seulement indiqué les régions où il se produit 1, 2 et 5 flammes froides. On a vu jusqu’à 8 flammes froides dans certains systèmes. Ces flammes sont incapables d’amorcer la combustion de papier et sont habituellement de couleur pâle (par exemple : bleu pâle), et leur analyse y révèle la présence de peroxydes et d’aldéhydes: l’émetteur principal de ces flammes est le formaldéhyde électroniquement excitée. Cette péninsule de stabilité est donc une zone de stabilité relative. Il y a oxydation lente de l’hydrocarbure : les réactions ramifiantes ne peuvent prendre leur importance. Cependant, le mélange réactionnel évolue lentement vers la formation des produits finaux. En enregistrant la variation de la pression, on peut suivre effectivement le déroulement lent mais efficace de cette oxydation : Fig. 7.6.
À chaque émission, il y a variation brutale de la concentration d’oxygène moléculaire, de l’oxyde de carbone et du propène. La concentration d’oxygène décroît, celles du CO et du propène augmentent. D’autres produits ont aussi des concentrations qui croissent : HCHO, CH3OH, CH3CHO, C2H4, acides, peroxydes,... Le mécanisme n’est cependant pas bien connu. Les parois du réacteur (forme et nature) interviennent également.
|
|
Figure 7.6. Variation de la pression dans un mélange C3H8 : O2 (1 : 1) à 280 °C.
L’étude de l’oxydation des hydrocarbures révèle que leur réaction d’oxydation ne donne lieu qu’à une seule limite d’explosion et que celle-ci a une nature principalement thermique (voir Fig. 7.3). Cependant, l’étude de cette figure révèle qu’un phénomène plus complexe se produit juste à l’extérieur de la limite d’explosion: il y a une péninsule d’explosion entre certaines limites de température. Cette situation est différente de celle de la réaction entre O2 et H2 où la région d’explosion est confinée entre deux limites de pression.
L’explication du déroulement de ce type particulier d’oxydation a été donnée en termes de ramifications dégénérées par SEMENOFF. Ce mécanisme implique la présence d’un processus par lequel se forme un produit intermédiaire, M, qui peut réagir pour donner, soit un radical, ou soit une molécule stable. Par exemple, dans certaines réactions, l’intermédiaire qui est un hydroperoxyde peut se décomposer pour donner les radicaux R• et •OH, ou, il peut donner une cétone et de l’eau :
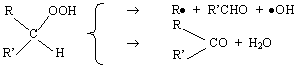
L’intermédiaire M ayant une vie assez longue, le processus de ramification est retardé : le nombre de radicaux s’accroît lentement, ce qui donne lieu à la présence d’une période d’induction. Au cours de l’oxydation, le nombre de radicaux augmente de façon telle en fonction du temps, qu’avant que la réaction devienne trop rapide (avant qu’il y ait véritable explosion), l’effet d’épuisement des radicaux est significatif et la réaction devient plus lente. Les flammes froides sont donc des explosions dégénérées de ce type: la vitesse est rapide, mais pas aussi rapide que celle d’une explosion classique. Ces explosions dégénérées sont du type thermique (elles sont dues à l’auto-échauffement).
Il y a une limite inférieure de température pour ces explosions parce que le processus de ramifications dégénérées diminue d’importance quand la température diminue, et qu’il devient trop lent pour que le nombre de radicaux puisse croître. La limite supérieure de température provient du fait qu’il y a alors un processus de rupture de chaîne qui prend de plus en plus d’importance quand la température augmente. C’est la raison pour laquelle ces vitesses d’oxydation ont des coefficients négatifs de température sous certaines conditions.
Aux conditions où le système se trouve dans la région de la péninsule d’explosion, il y a donc un domaine de température où la réaction a un coefficient de température négatif. À l’extérieur de ce domaine de température, la vitesse de la réaction augmente très rapidement, avec la température, de façon normale. La réaction principale qui consomme les hydrocarbures n’est pas ramifiée, mais un produit intermédiaire de la chaîne (l’agent de ramifications dégénérées), sous certaines conditions, induit une réaction secondaire, qui conduit à une ramification en chaîne. Si les énergies d’activation des réactions non ramifiées sont plus élevées que celles des réactions qui conduisent à la ramification, il y aura donc une région de température, où, l’accroissement de la température cause une réduction de la vitesse globale de la réaction (due au fait que la proportion des intermédiaires qui mènent à la ramification va alors diminuer).
Ainsi, l’instabilité thermique causée par cette variation inhabituelle de la vitesse par rapport à la température, peut donc mener à des écarts soudains de la température du mélange réactionnel à certains moments au cours de la réaction.
À titre d’exemple, on trouve ci-dessous le mécanisme simplifié suggéré par Walsh pour l’oxydation en phase gazeuse du n-propane.
| [1] | O2 + CH3CH2CH3 | ® | HO2 + CH3CH(•)CH3 | ||
| [2] | O2 + CH3CH(•)CH3 | ® |
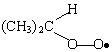 |
||
| [3] |
|
+ CH3CH2CH | ® |
|
+ CH3CH(•)CH |
| [4] |
|
® | CH3O• + HO• + CH3CHO | ||
| [5] | ® | (CH3)2CO + H2O | |||
| [6] | •CH3 + O2 | ® | CH3OO• | ||
| [7] | CH3OO• | ® | HCHO + HO• | ||
Le formaldéhyde produit à la réaction 7 pourra donner lieu à des réactions subséquentes. Il faut aussi noter que le radical (CH3)2CHOO• ne se décompose pas directement, mais il abstrait plutôt un atome d’hydrogène (réaction 3), produisant ainsi un hydroperoxyde qui donnera lieu à la ramification dégénérée.
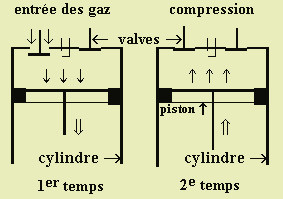
Le fonctionnement d’un moteur classique à quatre temps est le suivant. Dans un premier temps, l’ouverture d’une valve entraînée par l’arbre à cames permet l’admission dans la chambre du mélange combustible - comburant (essence - air) (Fig. 7.7. 1er temps). Dans un second temps, la remontée du piston comprime le mélange gazeux (Fig. 7.7. 2ème temps). Au sommet de sa course, la bougie apporte l’étincelle qui fait exploser le mélange : le piston est refoulé et l’énergie de la combustion entraîne le véhicule (Fig. 7.7. 3e temps). Finalement les gaz brûlés sont expulsés lors de la remontée du piston - quatrième temps (Fig. 7.7. 4e temps).
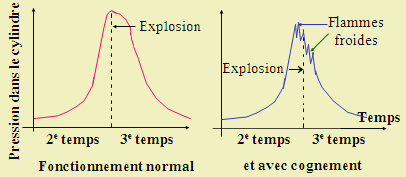
Les flammes froides peuvent apparaître vers la fin du deuxième temps, pendant la compression finale du mélange initial. Pendant la remontée du piston, la température et la pression du mélange augmentent. On entend alors ces bruits sourds : on dit que le moteur cogne. L’apparition des flammes froides est le résultat d’un mélange mal préparé ou d’une géométrie du piston mal calculée (Fig. 8).
Lorsque la bougie allume le mélange combustible - comburant (l'essence et l'air), le front de flamme se déplace rapidement dans le cylindre (plusieurs milliers de mètres à la seconde). Il arrive qu'avant même que la bougie étincelle, le mélange s'enflamme spontanément, donc au mauvais moment, réduisant la puissance récupérable. Pour prévenir et empêcher le cognement on utilise des essences à haut indice d'octane. Ce sont des mélanges contenant des hydrocarbures à chaîne ramifiée. On a défini une échelle d'indice d'octane entre 0 et 100. La valeur zéro pour cet indice correspond à une essence qui serait de l'heptane normal pur et la valeur 100 est arbitrairement attribué à l'isooctane pur (2,4,4-triméthylpentane). Avant 1971, on utilisait un additif le plomb tétraéthyle pour éliminer le cognement. Compte tenu des conséquences environnementales, ce produit a été banni. On l'a remplacé par le 1,2-dibromoéthane...
|
|
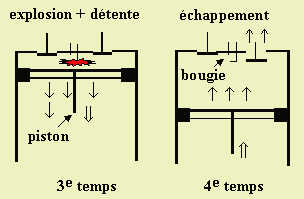 |
Figure 7.7. Schémas de description de fonctionnement du moteur à 4 temps.
(Voir une animation dans la section Diaporama, Ch. 7, diapos. n° 10).
|
|
Figure 7.8. Fonctionnement du moteur à combustion interne.
Variation de la pression dans le cylindre en fonction du temps (2e et 3e
temps).
5. L'émission de lumière par les flammes dynamiques
L’émission de lumière par un système en combustion peut provenir de plusieurs sources. Il y a l’émission du corps noir. Cette émission est liée seulement à la température de l’émetteur. Il y a aussi émission à partir de molécules excitées dans des états électroniques, principalement par fluorescence : voir Chapitre 5.
|
|
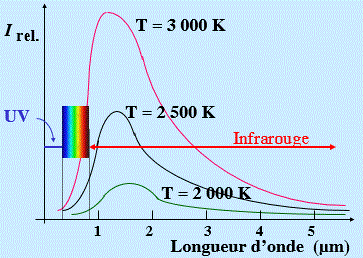
Figure 7.9. Émission du corps noir à diverses températures.
C’est l’émission normalement rencontrée par les braises dans un foyer. Cette émission correspond à l’énergie emmagasinée sous forme d’excitation de vibration dans le réseau cristallin. Cette émission existe à toute température. Cependant, à la température normale, elle est présente dans la région infrarouge et n’est donc pas visible. Elle se manifeste au-dessus de 1000 °C et s’accentue au fur et à mesure que la température s’élève : Fig. 7.9.
La chimiluminescence est très présente dans les flammes, bien qu’il existe des flammes très peu, voire même pas du tout, lumineuses. Dans la chimie complexe qui a lieu dans une flamme, de nombreuses espèces électroniquement excitées peuvent être formées par réactions chimiques : c’est la chimiluminescence. La flamme très jaune d’un feu de foyer peut être attribuée à l’émission des atomes de sodium (!) électroniquement excités (raies D). Ces atomes sont présents en assez grandes concentrations dans le bois, les bougies,...
H• + H• + Na ® H2 + Na*
H• + •OH + Na ® H2O + Na*
Na* ® Na + hn (raies D)
Le chalumeau oxyacétylénique produit une lumière à partir des radicaux hydroxyles rotationnellement excités. La température de cette flamme est de 3 400 K. La réaction suivante produit des radicaux ·OH animés d’un mouvement de rotation très important. On a pu montrer que sous une pression de 1,5 Torr, la température de rotation de ces radicaux est de 8 750 K. À la pression atmosphérique, où une certaine stabilisation (refroidissement) a lieu, la température de rotation est encore de 5 400 K. Il y a relaxation partielle à cette pression.
CH• + O2 ® •OHrot + CO
C2 + •OH ® CH* + CO
H2C-CºCH+ + e- ® CH3• + C2*
(CN)2 + O2 ® CNrot + ?
Dans ce dernier cas, la température rotationnelle est de 4 800 ± 200 K, alors que la température de la flamme (température macroscopique) est inférieure à 2 000 K.
Ils peuvent être nombreux: atomes, radicaux, molécules, ions,... Chaque flamme constitue évidemment un cas particulier. Les états excités à partir desquels ces entités peuvent fluorescer doivent avoir des temps de vie relativement courts. La théorie cinétique des gaz (Chapitre 2) prévoit que ces entités excitées peuvent être désactivées par collision. La vitesse de ces collisions est telle que :
![]()
Le symbole m représente la masse réduite. Dans les conditions TPN, une espèce A subit environ 8 109 collisions par seconde. Il faut donc que l’espèce excitée ait un temps de vie, t, inférieur à 10-9 s pour pouvoir émettre avant d’être stabilisée à la pression atmosphérique (t relatif à l’émission).
Tableau 7.1. Quelques émetteurs potentiels dans les flammes
| Émetteur | transition | l (nm) | t (seconde) |
| Na | 2S ® 2P | 589,0 - 589,6 | 1,6 10-8 |
| K | 2S ® 2P | 769,9 - 776,5 | 2,7 10-8 |
| Rb | 2S ® 2P | 794,7 - 780,0 | 2,8 10-8 |
| Li | 2S ® 2P | 670,8 | 2,7 10-8 |
| Hg | 1S ® 3P | 253,7 | 1,1 10-7 |
| Hg | 1S ® 1P | 184,9 | 1 10-9 |
| Tl | 2P ® 1S | 535 | 1,4 10-8 |
| •OH | 2S+ ® 2P | 306,4 | 1 10-6 |
| •CN | B2S+ ® x2S+ | 388,3 | 8,5 ± 1,0 10-8 |
| •CH | A2D ® x2P | 431,5 | 5,6 ± 0,6 10-7 |
| •CH | B2D ® x2P | 390,0 | 1,0 ± 0,4 10-7 |
| •NH | A3P ® x3S | 336,0 | 4,25 ± 0,6 10-7 |
Certaines de ces transitions sont normalement interdites (voir Chapitre 5). D’autres transitions ont été observées (Tableau 7.1) :
•CN à 359, 388,3 et 421,6 nm
C2 à 473,7 et 526,5 nm
NO• à 215,5, 226,9-247,9,... 259,5 nm
HCHO dans la région de 385-460 nm
C3 vers 400 nm, ...
|
|
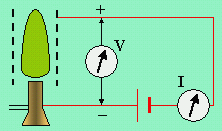
Figure 7.10. Schéma de
montage expérimental
montrant l’ionisation de flamme.
|
|
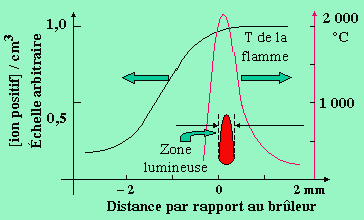
Figure 7.11.
Caractéristiques physicochimiques
d’une flamme.
6. L'ionisation des flammes
Dans le cas de la combustion des hydrocarbures, on observe la formation d’ions dans la flamme. En effet, si l’on entoure une flamme d’une grille métallique portée à un potentiel positif par rapport à la sortie du brûleur, on enregistre le passage d’un courant électrique indiquant la nature ionisée de la flamme (Fig. 7.6). En détaillant davantage cette étude, la distribution des ions et la température de la flamme apparaissent telles que représentées à la figure 7.7. On enregistre une concentration ionique maximum immédiatement au voisinage et après la zone lumineuse.
La réaction de chimie-ionisation probablement la plus efficace implique le radical ·CH. Elle est la suivante [Bayes, K. D., Chem. Phys. Lett., 152 (4,5), 424 (1988)] :
| •CH (x2P), état fondamental : | DH = + 83 kJ/mol |
| •CH (a4S) + O (3P) ® CHO+ + e- | DH = + 12 kJ/mol |
| •CH (A2D), état électronique excité : | DH = - 192 kJ/mol |
et aussi, bien qu’avec une probabilité moindre :
| •CH* + O2 | ® CHO+ + O + e- | DH = - 188 kJ/mol |
| •CH* + HO2· | ® CHO+ + OH- | DH = - 96 kJ/mol |
| •CH* + HO2· | ® H• + CHO2+ + e- | DH = + 8 kJ/mol |
Les notations x2P, a4S et A2D représentent des états électroniques différents du radical •CH. Cette propriété est utilisée pour la régulation des brûleurs. En effet, la production d’ions est maximum lorsque le mélange comburant : combustible est stœchiométrique. Il y a alors optimisation du fonctionnement. Au laboratoire, cette chimie-ionisation est à la base du détecteur à ionisation de flamme utilisé en chromatographie gazeuse. Comme le nombre d’ions générés est proportionnel au nombre d’atomes de carbone non-oxydés, on dispose ainsi d’un détecteur sensible et de calibrage simple.
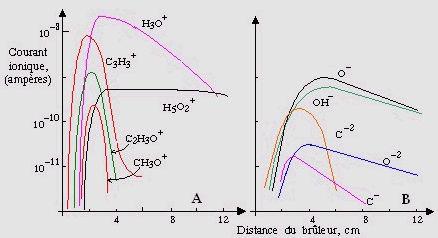
Ces ions positifs, qui seront traités en détail au chapitre 11, induisent des réactions de type ion - molécule. La cinétique en est complexe. Cependant, on a pu montrer que l’ion C3H3+ est l’un des ions (sinon l’ion) dont la concentration est la plus importante dans les premiers mm. À plus longue distance, donc loin de la flamme, l’ion H3O+ a la concentration la plus importante :
à faibles distances : [C3H3+] > [C2H3O+] > [C3HO+] > ...
à longues distances : [H3O+] > [H5O2+] > ...
|
|
Figures 7.12. Profils des ions positifs (A) et
négatifs (B) dans une flamme
d’acétylène/oxygène [H. F. Calcote et
al., 10 e Symp. Comb., Univ. Cambridge,
Cambridge, Angleterre, 605-619 (1965)].
Quant aux ions négatifs, les plus importants, en termes de concentrations, sont : [O-] @ [OH-] > [C2-] , etc.
e- + •OH + M ® OH- + M
O• + e- ® O- + hn
HO• + e- ® OH- + H• etc.
7. Formation de la suie ou du noir de carbone
Problème majeur dans l’encrassement des cheminées, contamination de l’environnement, mais aussi synthèse du noir de carbone pour la fabrication du cirage, pour la préparation de catalyseur (carbone activé), la combustion incomplète des hydrocarbures a reçu beaucoup d’attention, bien que les processus restent encore mal connus. L’étudiant a déjà vu la combustion incomplète des hydrocarbures aromatiques qui produit un abondant nuage noir. La combustion de la bougie permet de fumer une vitre qui, ainsi préparée, sert à regarder (avec quelques dangers) les éclipses solaires. Deux propositions de mécanismes ont été suggérées. Le premier met en cause des radicaux en C3 et le second l’oxydation partielle de l’oxyde de carbone.
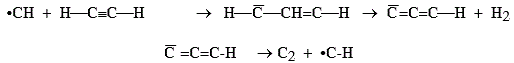
CO + •H ® C + •OH
Le tiret surmontant certains atomes de carbone spécifient la présence d’un doublet électronique disponible en périphérie de l’atome. D’autres mécanismes réactionnels ont aussi été proposés sans cependant être accompagnés de preuves certaines. Il a été remarqué que les ions C3H3+ jouent un rôle important dans les flammes: Fig. 7.6. On retrouve à nouveau une espèce qui à l’état radicalaire a déjà fait l’objet de soupçons importants quant à la formation des suies...
REMARQUE
À ce moment, il est pertinent de rappeler que le carbone, en plus de se retrouver sous les deux formes usuelles que sont le graphite et le diamant, peut aussi se retrouver sous la forme de "buckminsterfullerène". Alors que le diamant consiste en une structure tridimensionnelle très symétrique d'atomes de carbone tétragonaux (sp3), le graphite est constitué de feuillets de graphène empilés les uns sur les autres. Chaque feuillet est constitué d'une structure bidimensionnelle d'atomes de type sp2. Récemment, on a en effet observé que le carbone pouvait constituer de grosses molécules de formule C60, C70, ... La molécule C60, la plus stable, est synthétisée sous atmosphère d’hélium (150 mmHg) en effectuant une décharge électrique entre deux électrodes de charbon. Cette molécule a la forme d’un ballon de soccer (football anglais et français) et est soluble dans le benzène. Elle se retrouve également dans les fumées de combustion incomplète des hydrocarbures aromatiques.
Tableau 7.2. Différentes variétés de carbone
| Carbone | Densité | Indice de réfraction | Structure cristalline |
Arête de la maille élémentaire (nm) |
| g · dm-3 | ||||
| graphite | 2,09 - 2,23 | opaque | hexagonal compact | a = 0,2464 c = 0,6736 |
| diamant | 3,51 | 2,41 à 2,45 2,4175, lumière jaune du sodium |
cubique à faces centrées | 0,3567 |
| C60 | 1,7 | 2,2 à 630 nm | cubique à faces centrées | 1,4198 |
D'autres structures plus sophistiquées ont été mises en évidence. Par exemple, un feuillet de graphène peut s'enrouler sur lui-même et donner lieu à la formation d'un nanotube. Plusieurs feuillets peuvent aussi s'enrouler sur eux-mêmes et donner lieu à la formation de nanotubes imbriqués les uns dans les autres, à la manière de poupées russes, pour donner des nanotubes multi-parois ou multi-feuillets. Ces nanotubes ont des propriétés de surface très intéressantes puisque l'on peut y greffer des entités organiques ou inorganiques relativement stables, ce qui leur confère des propriétés catalytiques variées et importantes.
Conclusions
|
Problèmes
N° 1 Composition
Des
gaz de combustion ont la composition suivante (pourcentage en volume) :
CO2 :
10,5 %; CO : 1,1 %; O2 : 7,7 % et N2 : 80,7
%.
En
supposant que les gaz agissent comme des gaz parfaits, calculez :
1-
le pourcentage en masse des composants ;
2-
le volume d’un kg de gaz sous une pression de 740 mmHg et à la
température de 80 °C
et
la
densité du gaz en kg/m3 dans les mêmes conditions.
N° 2. Température de flamme
Calculez la température maximum des produits de combustion de la flamme issue d’un mélange stœchiométrique air – méthane lui-même à la température de 300 K et à la pression atmosphérique. On supposera que 90 % du méthane sont effectivement oxydés.
CH4 + 2 O2 ® CO2 + 2 H2O ; DH298 = 756,3 kJ
On donne les valeurs des capacités calorifiques suivantes (en J · K-1 · mol-1 entre 300 et 2000 °C) :
| CP(CH4, g) = 31,5 + 0,014 T | CP(CO2, g) = 32,35 + 0,005 T |
| CP(O2, g) = 26,0 + 0,0045 T | CP(H2O, g) = 34,2 + 0,002 T |
| CP(N2, g) = 28,3 + 0,003 T |
N° 3 Décomposition thermique de l'azométhane
La constante de vitesse du premier ordre de la décomposition thermique de l'azométhane est égale à :
kµ
= 1015,67 e-51000/RT
s-1
et la réaction est exothermique (DH
= -
43 000 cal · mol-1).
Des
études de cette décomposition ont été faites dans un réacteur dont le
rapport surface/volume est égal à 1 et dans des conditions où le coefficient
de transfert de chaleur était égal à
h
= 8,18 10-5
cal
·
cm-2
· deg-1
·
s-1
Calculer
la température du gaz immédiatement sous la limite d'explosion quand la température
du réacteur est de 360 °C. Calculer
aussi la pression de l'azométhane (en torr) à la limite d'explosion.
Quel serait l'effet sur la pression de l'azométhane si le diamètre du réacteur
est réduit ?
Pour en savoir un peu plus :
Ashmore, P. G., F. S. Dainton et T. M. Sugden, Photochem. and Reactions Kinetics, Cambridge University Press, 1967; QD601 P575.
Kondratev, V. N., Chemical Kinetics of Gas Reactions, traduction Pergamon Press, Oxford 1964; QD501 K82.
Gaydon, A. G. et H. G. Wolfhard, Flames, Chapman and Hall, ltd., 3e édition révisée, 1970.
Gas-Phase Combustion, Comprehensive Chem. Kinetics, vol. 17, édité par Bamford, C. H. et C. F. H. Tipper, Elsevier Sc. Publ. Co., New York, 1977; QD 501 C737 D296.
Huffman, D. R., Solid C60, Physics Today, 22, novembre 1991; QC23 P5783.
Pilling, M. J. et P. W. Seakins, Reaction Kinetics, Oxford Science Publ., 1996, 305 pages.
Sur les nanotubes, leurs structures et leurs propriétés : E. Gravel, D. Bernard, I. Namboothiri et É. Doris, "Nanotubes de carbone et catalyse hétérogène", L'Actualité chimique, 393-394, 82-88 (février-mars 2015).
Voir aussi, sur le même sujet : http://fr.wikipedia.org/wiki/Nanotube_de_carbone (site visité le 2020-11-06).
Anonyme, Un point sur "Le noir de carbone", L'actualité chimique, Société chimique de France, N° 395, 63-64 (avril 2015).
Sur le Net:
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes indusries, les Fondations, ...
Dernière mise à jour : 2020-11-06.
x x x x x x x x x x x x x x x
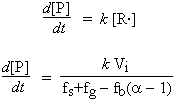
{kind=link}