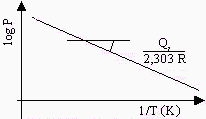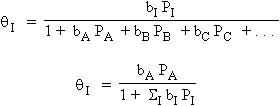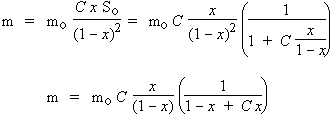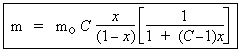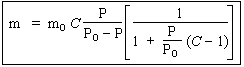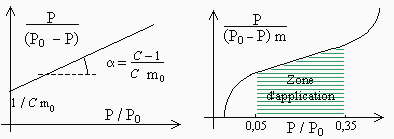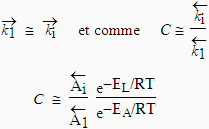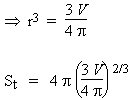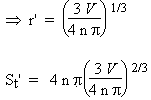|
|
CHAPITRE 12
LA CINÉTIQUE EN PHASE HÉTÉROGÈNE
|
Objectifs On a toujours considéré jusqu’à présent le cas de systèmes réactionnels ayant lieu au sein d’une phase homogène : la réaction s’y développant de manière généralisée au sein du réacteur.
|
Objectifs
spécifiques
-
Comprendre la nature et les spécificités des réactions mettant en jeu au
moins deux phases homogènes différentes.
-
Étudier différents cas d’espèce d’interactions entre des molécules
réactives et une surface de sorption physique ou chimique.
-
Être capable de calculer la surface active d’un catalyseur.
-
Comprendre et interpréter différents cas d’espèces de catalyse gaz solide,
liquide solide.
-
Approfondir la notion, la préparation, les propriétés, de catalyseur solides.
Les milieux réactionnels ne sont pas tous homogènes, bien au contraire. Comment se comportent les milieux hétérogènes par rapport aux milieux homogènes ? On peut penser que les principes développés pour la phase homogène sont encore valides. La simple présence d’au moins deux phases différentes, contenant chacune des réactifs différents, incite à penser que les problèmes de transfert de matière à travers la zone inter faciale interféreront grandement, peut-être même constitueront-ils l’étape limitante au déroulement de la réaction, imposant ainsi au système son allure réactionnelle. En outre, l’usage très fréquent de catalyseurs solides, dispersés au sein d’un milieu réactionnel, laisse présager des problèmes similaires liés aux propriétés de la zone inter faciale. En d’autres termes, les réactions physico-chimiques dans cette zone inter faciale sont appelées à jouer un rôle primordiale. Dans ces conditions, la notion de l’ordre est-elle encore significative ? On trouvera sans doute des expressions de vitesse sous la forme V = k [X]n... La molécularité n’aura plus rien de commun avec l’ordre.
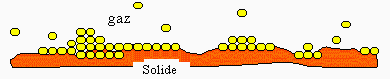
Puisque la zone inter faciale doit jouer un rôle primordial, voire prépondérant, en milieu hétérogène, il importe d’étudier les phénomènes de sorption. Par exemple, lorsqu’un gaz est au contact d’une substance métallique solide, que se passe-t-il à l’interface ? On a pu montrer qu’en passant de la phase gazeuse contenant les molécules G vers la phase du solide S, il y a une variation de la concentration des molécules G au fur et à mesure que l’observateur se rapproche de la surface du solide. Dans certains cas, le nombre de molécules G par unité de volume, se trouvant au contact de la surface du solide, est tel que l’on est en présence d’une phase liquide : G est littéralement condensé sur la surface du solide.
|
|
Figure 12.1. Zone inter faciale gaz-solide.
Dans le premier cas, la " condensation " du gaz sur la surface du solide est vraiment de nature physique. Il y a véritablement apparition d’une phase liquide, sans altération des molécules G dans cette phase.
Au contraire, en chimisorption, les molécules sorbées sont modifiées chimiquement. Ce peut être le cas de l’interface aluminium - oxygène. Dans cet interface, il se crée des molécules d’alumine Al2O3. Il n’est pas toujours facile de faire la différence entre les deux phénomènes. On peut d’ailleurs croire que la physisorption précède la chimisorption. Néanmoins, en mesurant les enthalpies des réactions de sorption on peut différencier les deux phénomènes (Tableau 12.1). On peut choisir arbitrairement une limite entre la physisorption et la chimisorption basée sur la chaleur de la réaction. Ce tableau permet de situer la frontière vers 40 kJ·mol-1. Note : Dans certains manuels on réserve le mot adsorption pour la sorption physique et absorption pour la sorption chimique.
Tableau 12.1. Exemples d’enthalpie de sorption
| Adsorbant | Molécule sorbée |
Physisorption Chimisorption (kJ·mol-1) |
T (°C) | |
| Fer | oxygène | 17 | 500 | - 183 |
| CO | 30 | 130 | -78 | |
| ZnO | hydrogène | 5 | - | - 191 |
| 85 | -200 | |||
| Cuivre | hydrogène | 4 | - | 78 |
| - | 90 | 160-300 | ||
Tableau 12.2. Adsorption de gaz sur 1 g de charbon activé à 15 °C
| Gaz | H2 | N2 | CO | CH4 | CO2 | HCl | H2S | NH3 | Cl2 | SO2 |
| V (cm3) TPN | 4,7 | 8,0 | 9,3 | 16,2 | 48 | 72 | 99 | 181 | 235 | 330 |
| T critique (K) | 33 | 126 | 134 | 190 | 304 | 324 | 373 | 406 | 417 | 430 |
Les quantités de gaz qui peuvent être adsorbées sur la surface d’un solide sont variables et dépendent bien sûr de divers paramètres dont la nature du couple gaz adsorbé-solide adsorbant. Le tableau 12.2 montre qu’il existe une relation étroite entre la quantité de gaz adsorbé exprimée en volume sous pression atmosphérique normales et la température critique du gaz adsorbé. Cet exemple montre que les quantités adsorbées par gramme de sorbant peuvent être importantes et justifient l’usage que l’on peut faire de cartouches de carbone activé dans certains types de masque à gaz.
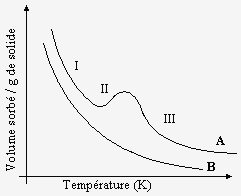
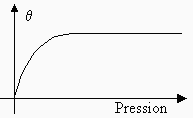
À pression constante, on représente la variation du degré d’adsorption en fonction de la température par une courbe qu’on appelle l’isobare d’adsorption. Dans ce cas aussi, la nature de l’adsorption influence la forme de la courbe (voir Fig. 12.2). Par exemple, dans le cas de l’adsorption de l’hydrogène sur du nickel (courbe A, Fig. 12.2), la première partie de l’isobare (partie I) représente la diminution normale de l’adsorption physique quand la température augmente. Par contre, la dernière partie de l’isobare (partie III) représente le phénomène d’adsorption chimique. Dans la région intermédiaire (partie II), la vitesse d’adsorption est plus lente, et la courbe est caractéristique du fait que l’adsorption chimique se fait en mettant en jeu une énergie d’activation non négligeable, dans ce cas.
|
|
Figure 12.2. Exemples d’isobares
d’adsorption : A: H2
sur nickel;
B: NH3 sur charbon de bois.
La chaleur d’adsorption
L’adsorption de molécules de gaz se traduisant par leur immobilisation (elles sont restreintes dans leurs mouvements), elle s’accompagne d’une diminution d’entropie. Comme l’adsorption implique aussi une diminution d’énergie libre, on peut conclure, à partir de la relation thermodynamique
D G = D H - TD S
que la chaleur d’adsorption sera elle-même négative. Ainsi, on peut dire que tous les processus d’adsorption sont exothermiques. Les processus d’adsorption étant des processus à l’équilibre, ils obéissent à une équation du type Clausius-Clapeyron :
| [1] |
|
Si Qa est définie comme étant la chaleur différentielle d’adsorption, l’équation précédente devient :
![]()
Comme à l’équilibre, le volume du gaz en phase gazeuse est beaucoup plus grand que le volume de gaz adsorbé, l’équation se réduit à :
![]()
Dans le cas où on a un gaz parfait, le volume d’une mole du gaz est donné par :
Vgaz = R T / P
Cette relation permet de modifier la précédente équation :
![]()
ou encore :
|
[2] |
|
L’intégration de cette équation permet de trouver :
| [3] |
|
La quantité Qa peut donc être déterminée à partir d’une droite représentant la variation de log P en fonction de 1/T (voir la Fig. 12.3). La chaleur d’adsorption, Qa qui peut être déterminée à partir de la dernière équation, est ce qu’on appelle la chaleur différentielle d’adsorption. Elle représente la quantité de chaleur par mole associée à un petit changement de recouvrement de la surface du solide quand le gaz s’y adsorbe.
|
|
Figure 12.3. Détermination de la chaleur différentielle d’adsorption.
La chaleur différentielle d’adsorption représente donc la chaleur dégagée par mole de gaz lors de l’adsorption menant à un petit changement de recouvrement de la surface du solide variant de q à q + d q, où, q est défini par :
![]()
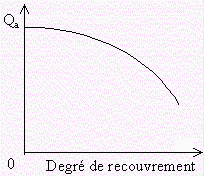
La chaleur d’adsorption différentielle dépend donc du taux de recouvrement de la surface du solide (Fig. 12.4).
La chaleur d’adsorption par mole, DHa correspond à l’adsorption d’une mole de gaz sur une quantité de solide telle que l’adsorption donne a mole de gaz par gramme de solide :
![]()
L’étude du phénomène de l’adsorption montre que la nature de celle-ci varie selon les situations étudiées. Dans certains cas, l’adsorption met en jeu des forces de liaison faibles, du type Van der Waals, similaires à celles qui sont impliquées lors d’une liquéfaction. On dit alors que ce phénomène est une adsorption physique ou physisorption. En général, l’adsorption physique se produit bien avant que le gaz n’atteigne une pression égale à sa tension de vapeur saturante, ce qui explique qu’elle se produit à des températures assez basses (voisines du point d’ébullition de la phase adsorbée). Elle met en jeu une chaleur d’adsorption assez faible (de l’ordre de 5 à 25 kJ mol-1), caractéristique du fait que la répartition des charges électroniques du gaz adsorbé est peu différente de ce qu’elle est normalement et que le gaz adsorbé a des propriétés à peu près identiques à celles des molécules libres. Elle est habituellement réversible et elle peut se faire de façon mono ou pluri moléculaire. Par contre, dans d’autres cas, l’adsorption met en jeu des énergies de liaison importantes. On dit alors qu’elle est une absorption chimique ou chimisorption. L’adsorption chimique résulte d’une profonde modification de la répartition des charges électroniques de la molécule adsorbée : les forces de liaison sont du même type que celles qui sont impliquées lors de la formation des liaisons chimiques. Elle met donc en jeu une chaleur d’adsorption assez grande, en général supérieure à 80 kJ mol-1, et elle se fait parfois avec une énergie d’activation. Elle est souvent irréversible (ou difficilement réversible) et elle se fait en une couche monomoléculaire.
|
|
Figure 12.4. Variation de la chaleur différentielle
d’adsorption
en fonction du degré de recouvrement de la surface de l’adsorbant.
La comparaison des deux types d’adsorption décrits ci-dessus, permet de croire que la physisorption ne présente pas beaucoup d’intérêt au point de vue de la catalyse. Par contre, dans le cas de la chimisorption, la chaleur d’adsorption est suffisamment élevée pour accélérer de façon notable la vitesse de certaines réactions. Il est donc concevable qu’elle puisse constituer l’étape intermédiaire de la catalyse hétérogène.
3. Les isothermes d'adsorption
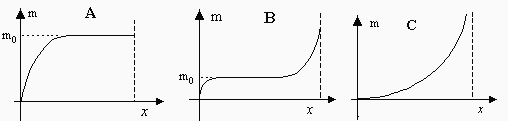
L’étude de l’adsorption d’un gaz sur la surface d’un solide montre qu’il y a formation d’un équilibre entre la pression de ce gaz et la quantité de gaz adsorbé par unité de masse de solide. On observe ainsi au moins trois types d’isothermes d’adsorption : Fig. 12.5. Dans le premier cas, Fig. 12.5 A, la quantité exprimée en masse de gaz adsorbé croît très vite en fonction de la pression. Elle atteint un plateau, comme s’il y avait saturation de la surface du solide. Autrement dit, le solide est complètement recouvert d’un film monomoléculaire liquide de molécules du gaz (Fig. 12.1). Bien sûr, la manipulation est limitée à la pression de vapeur saturante du gaz. Au-dessus de la pression de vapeur saturante, on provoque la liquéfaction du gaz. Le deuxième cas, Fig. 12.5 B, correspond au cas de figure précédent, avec en plus, après saturation de la surface du solide par une première couche, apparition de formation de plusieurs couches de molécules gazeuses sur la surface. Le troisième cas, Fig. 12.5 C, correspond à la formation de plusieurs monocouches sur la surface du solide, ou encore à la partie située à droite de la figure 12.5 B.
|
|
Figures 12.5. Isothermes d’adsorption sur un solide : 3 cas
typiques
x = P / Po; P : pression à l’équilibre; Po
: pression de vapeur saturante;
m : masse de produit sorbé par unité de matériau
sorbant.
Le développement de la représentation de LANGMUIR, pour une isotherme d’adsorption chimique, repose sur un certain nombre d’hypothèses :
![]()
La lettre G représente une molécule de gaz et S représente un site d’adsorption.
|
|
Figure 12.6. Représentation schématique du recouvrement monomoléculaire d'une surface.
Soient S la surface du solide et q la fraction de ce solide recouvert par des molécules. La vitesse d’adsorption est proportionnelle à la surface du solide inoccupé et à la pression du gaz. La vitesse de désorption est proportionnelle à la surface occupée. Puisqu’il y a équilibre entre les molécules en phase gazeuse et celles qui sont sorbées, on peut écrire :
vitesse d’adsorption = vitesse de désorption |
et :
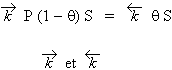
représentent les constantes de vitesse de sorption et de désorption. Comme dans le cas des constantes de vitesse de réaction, ils comprennent le facteur température. L’équation précédente se réarrange :
| [4] |
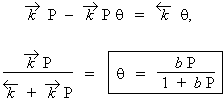 |
| [5] |
|
dans cette équation b est le coefficient d’adsorption : |
Ces résultats sont traduits par les figures qui suivent.
|
|
Figure 12.7. Représentation de l’isotherme
de LANGMUIR.
|
|
Figure 12.8. Représentation linéaire de l’isotherme
de LANGMUIR.
Si la pression P est très petite :
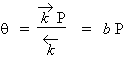
La lettre b représente la pente à l’origine de la courbe q = ƒ(P) : Fig. 12.7. Si au contraire, la pression P est très grande:
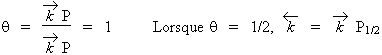
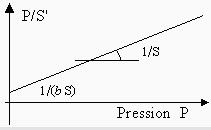
À une pression quelconque, il est préférable de transformer la fonction hyperbolique q = ƒ(P) en une fonction linéaire. Comme q est la fraction de la surface occupée S', et comme S est la surface totale,
|
[6] |
|
En multipliant par P et en divisant par S l'équation de droite, on obtient :
|
[7] |
|
La fonction P/S' = ƒ(P) est une droite d’origine 1/(b S) et de pente 1/S (Fig. 12.8).
Dans le cas où il y a plus d’un gaz qui peut s’adsorber sur le même solide, la théorie de Langmuir peut aussi être appliquée. Considérons le cas où l’adsorption donne lieu à une compétition entre un gaz A, de coefficient d’adsorption KA et un gaz B, de coefficient d’adsorption KB.

Dans le cas de l’adsorption concurrente de deux gaz sur une même surface solide, on peut poser :
q A =
fraction de la surface unitaire recouverte par le composé A
q B = fraction de
la surface unitaire recouverte par le composé B
q = fraction de la surface
unitaire non recouverte (libre).
On a bien sûr : q = 1 - (q A + q B)
À l’équilibre, on a :
| Vitesse
d’adsorption de A
= |
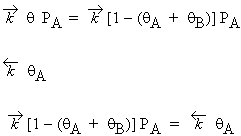 |
| Vitesse de désorption de A = | |
|
|
De cette équation on obtient :
|
[8] |
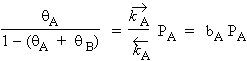 |
De même façon, on peut trouver :
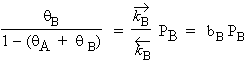
À partir de ces deux équations, on a :
|
[9] |
À partir de cette dernière équation, on trouve respectivement pour les deux équations qui suivent :
|
[10] |
Lorsque le produit bB PB est petit devant le produit bA PA, la première de ces deux équations se réduit à celle générale déjà vue : équation [5].
Il en est de même pour la seconde équation quand une situation analogue mais inverse se produit pour le gaz A.
L’extension de l’équation d’état de Langmuir peut aussi être faite pour un mélange de plusieurs gaz, A, B, C, ... Dans ce cas, la fraction de surface unitaire recouverte par n’importe lequel des gaz I, q I , est donnée par :
|
[11] |
|
|
ou encore, |
La présence de chacun des gaz se traduit donc par l’apparition d’un terme, qui lui correspond, au dénominateur des expressions représentant les fractions de recouvrement de chacun d’eux. Il en résulte que la fraction couverte par un gaz donné se trouve réduite par l’action des autres gaz. Aux pressions élevées ou en phase liquide, la saturation de la surface est pratiquement complète. Dans ce cas, la fraction de la surface couverte par chacun des gaz s’exprime alors par :
![]()
Comme la phase adsorbée joue un rôle d’intermédiaire en catalyse hétérogène, l’équation d’état de Langmuir, représentant la fraction de surface couverte par cette phase, qI, prend une très grande place dans la formulation cinétique du phénomène de catalyse.
Il arrive parfois que le processus d’adsorption s’accompagne de la dissociation en surface de la molécule adsorbée. Par exemple, il a été montré que l’adsorption de l’hydrogène, sur plusieurs métaux, se fait par l’adsorption d’atomes d’hydrogène, dont chacun occupe alors un site d’adsorption :
![]()
Le processus d’adsorption doit alors être considéré comme une réaction entre la molécule de gaz et deux sites d’adsorption. La vitesse d’adsorption est ainsi donnée par :
| Vitesse d'adsorption = | |
alors que
| Vitesse de désorption = |
|
À l’équilibre, on trouve :
Vitesse d’adsorption = Vitesse de désorption
![]()
D’où :
|
[13] |
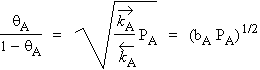 |
Ce qui donne :
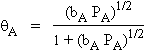
Quand la pression est suffisamment faible, ou quand le gaz est peu adsorbé :
(bA PA)1/2 << 1
On trouve alors :
q A = bA1/2 PA1/2
La fraction de la surface recouverte par le gaz adsorbé est alors proportionnelle à PA1/2. Par contre, quand la pression est grande, ou quand le gaz est fortement adsorbé :
(bA PA)1/2 >> 1
On montre que :
![]()
La fraction de la surface non couverte est alors inversement proportionnelle à PA1/2.
L’hypothèse de BET repose sur la formation de multicouches. Les molécules se posent les unes sur les autres pour donner une zone inter faciale qui peut contenir plusieurs épaisseurs de molécules sorbées : Fig. 12.9.
|
|
Figure 12.9. Représentation schématique de l'hypothèse des multicouches.
Posons
S0 la portion de surface inoccupée du solide
S1 la portion de surface occupée par une couche d’adsorbat
S2 la portion de surface occupée par deux couches d’adsorbat . . .
Si la portion de surface occupée par i couches d’adsorbat . . .
Sn la portion de surface occupée par n couches d’adsorbat . . .
Sur chaque couche, il y a équilibre dynamique entre le nombre de molécules qui s’adsorbent et celles qui se désorbent. On peut donc, sur chaque couche à l’équilibre, admettre que la surface demeure constante. Par exemple, la couche d’ordre 2 se forme par :
- sorbtion sur la couche d’ordre un + la désorption de la couche d’ordre 3.
La disparition de cette couche d’ordre 2 se fait par :
- désorption de cette couche + la formation de la troisième couche. Soit,
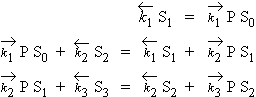 |
sur la couche d’ordre zéro |
| sur la couche d’ordre 1 | |
| sur la couche d’ordre 2, etc. |
En comparant les deux premières équations d’équilibre, on obtient :

En généralisant,
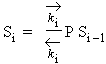
À partir de i = 2 et pour i > 1, il s’agit toujours d’une condensation de molécules du gaz sur les mêmes molécules et non sur la surface du solide. Donc :
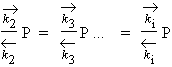
Posons ce rapport égal à x. On obtient ainsi :
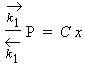
La constante C en est une de proportionnalité. Il vient :
S1 = C x S0
S2 = x S1 = C x2 S0
S3 = x S2 = x2 S1= C x3 S0 . . .
Si = x Si-1 = C xi S0 etc.
La surface totale du solide est telle que (en la ramenant à l’unité) :
1 = S0 + S1 + S2 + S3 + ... Si ... + Sn
1 = S0 + C x S0 + C x2 S0 + C x3 S0 + ... C xi S0 ... + etc.
1 = S0 [ C x + C x2 + C x3 + ... C xi ... + C xn ]
1 = S0 [ 1 + C (x + x2 + x3 + ... xi ... + xn )]
Posons  = x + x2 + x3 + ... xi ... + xn
Þ Â - x = x2 + x3 + ... xi ... + xn
|
x  -  = - x; Þ |
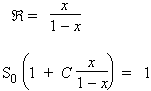 |
|
D’où : |
|
|
[14] |
Or x n’est évidemment pas accessible directement, ni d’ailleurs S0, S1, S2, ... Si, ... Sn. Soient m la masse totale de produit adsorbé et mo la masse de totale de produit nécessaire pour recouvrir le solide d’une seule monocouche.
m = m0 (0 S0 + 1 S1 + 2 S2 + 3 S3 + ... i Si + ... + n Sn)
m = m0 ( C x S0 + 2 C x2 S0 + 3 C x3 S0 + ... i C xi S0 + ... + n C xn S0)
m = m0 C x S0 ( 1 + 2 x + 3 x2 + ... + i xi-1 + ... + n xn-1)
Dérivons l'expression de  établie quelques lignes plus haut. Il vient :
d  / dx = 1 + 2 x + 3 x2 + ... + i xi-1 + ... + n xn-1
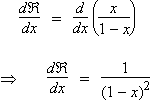
Donc :
et
|
[15] |
|
Si x tend vers 1, m tend vers l’infini, m ® ¥ , ceci ne peut être vrai que pour P = Po. Donc, si x = 1
| Þ | 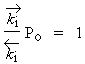 |
| Donc, | 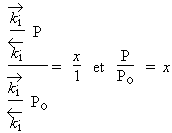 |
Sauf au point de liquéfaction, x est toujours inférieur à l’unité et est égal à la pression réduite du gaz.
|
[16] |
|
Si la pression P est faible, x l’est aussi et devient faible devant l’unité ou encore si P << Po, x << 1
|
[17] |
|
|
où m = mo |
|
équation dans laquelle :
A = C / Po et B = (C - 1) / Po
On retrouve la formule de LANGMUIR. En effet, à basse pression, l’approche de BET suppose que seule la première couche est en formation. Cela correspond à l’hypothèse de LANGMUIR. Cette dernière formule est donc un cas limite de la formulation de BET.
On représente à l’occasion le phénomène d’adsorption selon ce qu’on appelle l’isotherme de FREUNDLICH. La fraction de la surface occupée par un composé A est donnée par la relation
|
[18] |
qA = q bA PA1/n |
On a d’ailleurs vu plus haut que l’absorption de l’hydrogène sur certains métaux rejoint cette formulation. La figure suivante résume l’ensemble des expressions de qA en fonction de la pression. Les isothermes de FREUNDLICH et de LANGMUIR s’appliquent à la partie incurvée de la courbe qA = f(q). Les deux asymptotes correspondent aux cas limites.
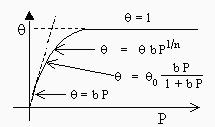 |
Figure 12.9.
Représentations de LANGMUIR et de FREUNDLICH
de l’adsorption.
4. Mesure des aires spécifiques
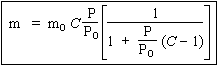
| L’isotherme : |
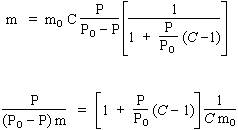 |
|
peut se réécrire sous la forme : |
En traçant le graphe
![]()
on doit obtenir une droite dont l’ordonnée à l’origine et la pente sont donnés par (Fig. 12.10) :
- ordonnée à l’origine : 1 / C mo
- pente à la droite : C - 1 / C mo
|
|
Figures 12.10. Représentations linéaire et réelle de l’isotherme de BET.
En réalité, on obtient généralement une bonne linéarité seulement quand la valeur de x est telle que : 0,05 < x < 0,35. À l’aide de cette ordonnée à l’origine et de la pente, on a deux équations dont les inconnues sont C et mo. On peut donc atteindre la connaissance de ces deux paramètres.
La théorie de BET prévoit la possibilité d’une infinité de couches. Lorsque le solide est très poreux, il suffit parfois de quelques couches de molécules pour remplir les alvéoles. Tout se passe comme si après l’adsorption de ces quelques couches, la surface du solide se rapetissait, expliquant ainsi la courbure observée pour des valeurs de x > 0,35.
Il faut mentionner, puisqu’il y a proportionnalité entre la masse et le volume de gaz correspondant, que l’on peut substituer, dans les formules précédentes, m et mo par V et Vo.
![]()
Connaissant la dimension d’une molécule, à l’aide de V, donc de sa surface, on saura déterminer l’aire spécifique de la surface du solide.
5. Mesure de la chaleur d'adsorption
C est une constante qui dépend de la température. En effet :
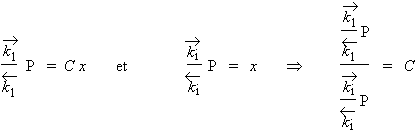
Or les constantes
![]()
sont gouvernées par la vitesse des collisions des molécules de la phase gazeuse sur la paroi inoccupée et occupée du solide. Donc,
|
[19] |
|
En admettant que
![]()
sont voisins (il s’agit du passage de molécules condensées vers la phase gazeuse) :
|
[20] |
|
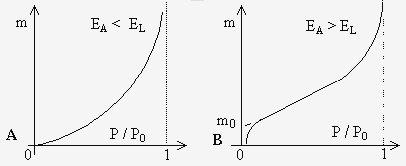
EA = chaleur
d’absorption de la première couche et
EL = chaleur de liquéfaction du gaz, donc de la formation des couches
supérieures. Si EA < EL , on obtient l’isotherme
décrit en figure 12.11 A. Si au contraire, EA > EL
, on obtient celui décrit à la figure 12.11 B.
|
|
Figures 12.11. Isothermes de BET selon les valeurs relatives des chaleurs d'adsorption et de liquéfaction.
6. Préparation des catalyseurs solides
Comme on le voit, la surface active d’un catalyseur doit être la plus grande possible pour optimiser l’efficacité des catalyseurs solides. Il doit donc se présenter sous la forme d’une poudre la plus finement divisée. Comparons la surface disponible d’un volume V d’un solide se présentant sous la forme d’une seule boule ou sous la forme de n billes :
| a- division en 1 seule boule : |
b- division en n billes : |
|
|
V = 4/3 p n r'3 |
|
|
|
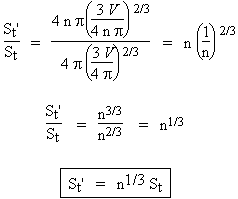
La surface du catalyseur croît comme la puissance 1/3 de n. Une division de la boule initiale par un facteur de 1 000 entraîne une augmentation de 10 fois de la surface. Il y a donc intérêt à obtenir un catalyseur le plus finement divisé. Une façon de le faire consiste à réduire un oxyde métallique par de l’hydrogène. Pourvu que l’oxyde soit relativement divisé pour obtenir une meilleure réduction, celle-ci forme un métal extrêmement poreux. On obtient ainsi des surfaces actives qui peuvent dépasser plus de 100 m2 par gramme de solide pulvérulent. Plus récemment, le domaine des nanomatériaux est venu ouvrir des perspectives intéressantes dans le développement de nouveaux, sinon catalyseurs, à tout le moins à de nouveaux modes de leur préparation.
7. Adsorption de gaz par des soldes
Soit la décomposition d’un composé gazeux A sur la surface d’un catalyseur et supposons que la réaction la plus lente soit le processus d’adsorption. Supposons enfin que la pression du gaz soit très inférieure à sa pression de vapeur saturante.
A (g) ® B, avec P(A) < < Po(A)
La vitesse de réaction est gouvernée par l’étape la plus lente. De plus, comme la pression est faible, la surface du catalyseur est partiellement et faiblement recouverte :
qA = b P,
Vréact.
=
k'
qA
= -
dP / dt
=
k'
b P
= k
P
Selon l’isotherme de LANGMUIR :
| Cette équation se réécrit : |
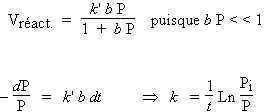 |
Cette équation est identique à ce que l’on a obtenu pour une réaction d’ordre 1. Ce résultat est compréhensible puisque l’étape limitante est gouvernée par le processus d’adsorption lui-même contrôlé par la pression du gaz. On dira que k est la constante de vitesse de réaction hétérogène d’ordre 1. On connaît beaucoup de réactions de ce type.
Les décompositions de :
- l’oxyde nitreux sur l’or : N2O / Au
- l’iodure d’hydrogène sur le platine : HI / Pt
- l’acide formique sur divers catalyseurs : HCOOH / SiO2, Pt, Rh,...
- la phosphine sur la silice : PH3 / SiO2,...etc.
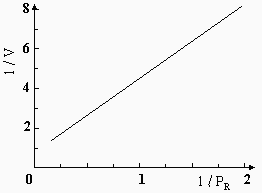
En supposant qu’il n’y a que le réactif, R, qui est fortement adsorbé alors que le produit P ne l’est pas, selon l’isotherme de LANGMUIR, la vitesse de la réaction se résume à
|
[21] |
|
La grandeur V représente la vitesse de la réaction. Dans ce cas, la réaction observée ne correspond pas à une réaction simple : l’expression de sa vitesse n’étant pas un simple produit de termes de concentration, elle n’a pas d’ordre caractéristique. On peut cependant vérifier si une réaction obéit à cette équation en la mettant sous la forme :
![]()
Si la réaction observée est conforme à l’équation, l’inverse de sa vitesse doit être une fonction linéaire de l’inverse de sa pression. Ce comportement se vérifie pour beaucoup de réactions, comme par exemple, pour celle de la déshydrogénation du cyclohexane sur catalyseur de Cr2O3 (Fig. 12.12) :
|
|
Figure 12.12. Déshydrogénation du cyclohexane catalysée par Cr2O3.
Dans le cas limite où q = 1 la réaction est indépendante de la pression dans le réacteur et
- d P / dt = k
| L’intégration donne : k = | |
On obtient une réaction d’ordre zéro : la vitesse de la réaction ne dépend pas de la concentration du réactif.
Exemples de tels cas, les décompositions de
l’ammoniac sur le tungstène : NH3 / W
l’ammoniac sur le molybdène, l’osmium : NH3 / Mo, NH3 / Os,...
l’iodure d’hydrogène sur l’or : HI / Au, .... etc.
Dans ce cas intermédiaire entre les deux précédents :
![]()
Cette dernière équation est souvent remplacée par l’isotherme de FREUNDLICH :
|
- dP / dt = k Pn |
La réaction est alors d’ordre n. |
1er cas : soit la réaction A ® B + C, où A est faiblement adsorbé et où l’un des produits, par exemple B, s’adsorbe très fortement. Il empêche ainsi A de s’adsorber. Il y a donc ralentissement au fur et à mesure que la réaction progresse :
- d PA / dt = k' (1 - qB) PA
Le symbole qB est la portion de surface occupée par le produit B. La surface disponible pour la catalyse est évidemment proportionnelle au facteur (1 - qB). Si qB tend vers 1,
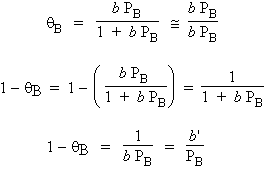 |
= 1 et b PB > > 1 |
| et puisque b PB > > 1 | |
|
avec b' = 1 / b |
La fraction de la surface libre est inversement proportionnelle à la pression de B :
C’est le cas de la décomposition de l’ammoniac sur le platine à 1138 °C, NH3 / Pt. L’azote est sans effet. Cependant l’hydrogène libéré s’adsorbe sur le platine et occupe ainsi une partie de la place qui devient non disponible pour la réaction de décomposition. Dans le cas de la décomposition de l’oxyde nitreux sur le platine, N2O / Pt, ou sur l’oxyde de cadmium, N2O / CdO, l’oxygène libéré est modérément adsorbé : on dira aussi que l’un des produits, ici l’oxygène, est un agent retardateur.
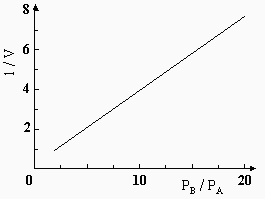
2ème cas : Soit la réaction A ® B + C, où le réactif A est fortement adsorbé de même que l’un des produits, par exemple B. Ce dernier empêche ainsi A de s’adsorber. Il y a encore ralentissement au fur et à mesure que la réaction progresse. L’équation de vitesse devient :
|
[25] |
|
On peut simplifier cette équation quand la réaction se fait à pression suffisamment haute ou en phase liquide. Dans ces conditions, la sommation bA PA + bB PB du dénominateur de l’équation est beaucoup plus grande que 1, ce qui donne alors :
![]()
Si on inverse l’équation et qu’on pose V comme étant la vitesse de la réaction on a :
|
[26] |
|
En portant en graphique 1/V en fonction du rapport PB /PA, on obtient une droite dont l’ordonnée à l’origine est 1/ k' et dont la pente permet de mesurer le rapport des coefficients d’adsorption. Par exemple, les réactions d’oxydation des alcools secondaires en cétones sur catalyseur de nickel obéissent à cette loi (Fig. 12.13).
|
R-CHOH-R' |
|
RCOR' + H2 |
|
|
Figure 12.13. Tracé selon
l’équation obtenue pour la
déshydrogénation
du cyclohexanol à 180 °C.
Cette situation se trouve quand une substance autre que le réactif s’adsorbe fortement à la surface du catalyseur (ce peut être soit une substance étrangère, soit un produit secondaire, etc.). L’équation de vitesse devient alors :
|
[27] |
|
Le produit bE PE est relatif au corps étranger. Dans le cas où la pression du réactif serait faible, l’équation devient :
![]()
Si l’inhibiteur est fortement adsorbé (bE PE >> 1), cette équation se réduit à
![]()
. La vitesse de la réaction globale est alors du premier ordre par rapport au réactif et, comme il se doit, elle est inversement proportionnelle à la pression de l’inhibiteur (la présence de l’inhibiteur ralentit la réaction).
8. Adsorption de liquide par des solides
Un cas très fréquent d’une telle adsorption se rencontre avec les solides cristallins qui adsorbent les ions, avec le charbon activé qui adsorbe les non-électrolytes. La théorie de tels systèmes est moins facile et le plus souvent les traitements expérimentaux ont imposé leur traitement mathématique. En général, ces cas sont représentés par l’isotherme de FREUNDLICH :
y = k C1/n |
où :
y est la masse de substance adsorbée par unité de masse adsorbante
C est la concentration du matériel adsorbé à l’équilibre en solution
k et n sont des constantes qui dépendent du système, de la température...
Þ Ln
y = Ln
k + 1/n Ln C
Exemple : adsorption de l’acide acétique sur du charbon activé (charcoal) :
y = 0,160 C 1/2,32
La catalyse ne demande pas d’identification d’exemples précis : ils sont extrêmement nombreux (Tableau 12.3) dans le domaine industriel, biochimique,... Il importe cependant de rappeler certaines applications de la sorption. De petits systèmes hermétiquement fermés utilisent les propriétés d’adsorption pour l’obtention du vide. Une enceinte préalablement évacuée est scellée sous vide. Elle contient un matériau adsorbant comme du charbon activé. Placée à la température de l’azote liquide (- 193 °C), les molécules qui se seront dégazées des parois de l’enceinte seront adsorbées sur le charbon activé, créant ainsi un meilleur vide.
La purification de mélanges gazeux. Le tableau 12.2 montre qu’il est, par exemple, facile d’éliminer des traces de SO2 dans un réservoir d’hydrogène,...
L’analyse par la chromatographie en phase gazeuse ou en phase liquide utilise les propriétés de la physisorption....
Un autre aspect intéressant de la catalyse est la diversité des réactions que l’on peut générer suite à un choix judicieux du catalyseur et des conditions opératoires. Il est bien connu que l’alumine est un excellent catalyseur de déshydratation alors que les métaux de transition sont plutôt des catalyseurs de déshydrogénation :
| 4 NH3 + n O2 | ® 4 NO + 6 H2O en présence de platine |
| ® 2 N2 + 6 H2O en présence de nickel |
Tableau 12.3. Exemples de réactions industrielles catalysées
| Réaction impliquée |
Catalyseur |
| Déshydrogénation | |
| C6H5-CH2-CH3 ® C6H5-CH=CH2 | Fe2O3 avec Cr2O3 et K2CO3 |
| CH4 + H2O ® CO + 3 H2 | Ni |
| alcool benzylique ® benzaldéhyde | NTSC* recouvert de Au |
| Hydrogénation | |
| N2 + 3 H2 ® 2 NH3 | Fe avec Al2O3 + K2O + MgO |
| C6H6 + 3 H2 ® C6H12 | Ni ou Pt |
| 3-méthyl-2-buénal + H2® 3-méthyl-2-buténol | NTSC* recouvert de Pt |
| Oxydation | |
| 2 SO2 + O2 ® 2 SO3 | V2O5 et K4P2O7 |
| 4 NH3 + 5 O2 ® 4 NO + 6 H2O | Pt-Rh sur toile métallique |
| 2 CH2=CH2 + O2 ® oxyde d'éthylène | Ag sur support |
| C6H6
+ 4,5 O2
+
2 CO2
® anhydride malique + 6 H2O |
V2O5 + MoO3 |
| alcool benzylique (+ H2O) ® acide benzoïque | NTSC* recouvert d'or. |
| Désulfuration | |
| SO2 + 2 H2S ® 3 S + 2 H3O | Al2O3 |
|
Tiré en partie de Chimie industrielle, Perrin, R. et J.-P. Scharff, 2e édition, Masson, Paris, 1997. |
|
| *: NTSC = nanotube simple de carbone. L'actualité chimique, 393-394, 82-88 (février-mars 2015). | |
- soit la décomposition catalytique de l’éthanol et de l’acide formique :
| C2H5OH | ® C2H4 + 4 H2O en présence d’alumine |
| ® CO + H2O en présence de Cu et de Ni | |
| HCOOH | ® CO + H2O en présence d’alumine |
|
®
CO2
+ H2
en présence de cuivre |
Tableau 12.4. Polymérisation catalysée du 1,3-butadiène
| Catalyseur | Structure du polybutadiène obtenu |
| TiCl4 + (C2H5)3Al | cis-poly-1,4-butadiène |
| TiCl4 + (C2H5)3Al à 25 °C | trans-poly-1,4-butadiène |
| Cr(CO)6 + (C2H5)3Al à 65 °C | butadiène poly(isotactique-1,2) |
| Cr(CO)6 + (C2H5)3Al | butadiène polysyndiotactique |
|
|
|
| polymère isotactique | polymère syndiotactique |
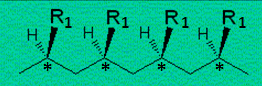
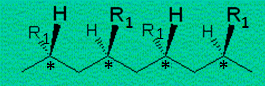
Figures 12.9. et 12.10. Deux structures du polybutadiène (R º •CH=CH2).
Les carbones asymétriques,
C*, sont tous en configuration R ou en configuration S dans les polymères
isotactiques : les groupes alkyles, aryles, halogénés, ... R1 sont du même côté de la chaîne hydrocarbonée.
Par contre, les carbones* sont alternés, R et S, dans le polymère syndiotactique : les groupes R1 sont alternativement d'un côté et de
l'autre de la chaîne. En polymérisation radicalaire ou
anionique, les groupes R1 sont
distribués au hasard d'un côté ou de l'autre de la chaîne: on
obtient un polymère de structure atactique.
Bien évidemment,
les propriétés physiques, mécaniques,... de ces polymères dépendent de la
structure moléculaire et varient donc d'un polymère à l'autre.
Les mêmes réactions de polymérisation avec les différentes structures peuvent
être obtenues avec le propène (R º •CH3)
ou avec le styrène (R º
•C6H5).
Tableau 12.5. Hydrogénation catalysée de l’oxyde de carbone
| Réactifs | Catalyseur et conditions | Produits |
| x CO + y H2 | ZnO seul à 400 °C et 200 atm | CH3OH |
| ZnO légèrement alcalin | CH3OH + isobutanol | |
| ZnO + Fe2O3 | CH4 + homologues sup | |
| Co (réduit) à 200 °C et 200 atm | hydrocarbures supérieurs | |
| CH4 |
10. Conclusions
|
loi d’ordre 1. Hinshelwood a mesuré une vitesse de 5,5 10-4 s-1 à 140 °C et de 9,2 10-4 s-1 à 185 °C.
Calculez l’énergie d’activation de cette réaction.
Quelle est l'aire d'un cube ayant 1 cm de côté ? Quelle
serait l'aire totale obtenue si ce cube était subdivisé en petits cubes de
grandeur colloïdale, c'est-à-dire en petits cubes de 10-7 cm de
côté ? Exprimer le résultat en hectare.
Des mesures de l'adsorption du CS2 sur un solide
ont été faites de -10 à +30 °C. À l'aide des données expérimentales
fournies au tableau, déterminer la chaleur différentielle d'adsorption du CS2
sur ce solide pour différentes concentrations de surface du CS2
(i.e. pour différentes valeurs de X données au tableau). Cette adsorption
est-elle physique ou chimique ?
Langmuir a trouvé les valeurs suivantes pour l'adsorption du CH4 sur du mica à 90 K : X représente le volume de gaz TPN adsorbé sur le solide en équilibre avec la pression de ce gaz.
|
Pression (bar) |
X (cm3 g-1) |
|
13,4 |
85,0 |
|
11,1 |
80,4 |
|
9,60 |
75,9 |
|
8,55 |
71,6 |
|
7,40 |
67,9 |
|
6,88 |
64,2 |
|
5,85 |
61,2 |
À l'aide de ces données, vérifier graphiquement la validité de l'isotherme de Langmuir pour ce système. Calculer ensuite le coefficient d'adsorption du CH4 sur le mica.
L'adsorption du CO2 sur du charbon de bois a été
mesurée à 0 °C pour différentes pressions. Employez l'isotherme
d'adsorption de Langmuir pour trouver l'aire spécifique du charbon de bois si
l'aire occupée par une molécule est 0,141
nm2.
|
Pression (mmHg) |
X (cm3·g-1) |
|
27,0 |
19,3 |
|
135 |
58,8 |
|
167 |
64,7 |
|
249 |
76,4 |
|
291 |
80,6 |
En 1930, McBain et Britton ont
étudié l’adsorption de l’éthylène sur du charbon activé à 273 K. Montrez, en
utilisant un graphe convenable, que leurs résultats s’accordent avec l’isotherme de
Langmuir. De ce graphe, calculez la valeur ka / kb
(où ka et kb sont, respectivement, les constantes de
vitesse d’adsorption et de désorption) ainsi que la surface active d’un gramme de ce
charbon. On rappelle que la surface de section d’une molécule d’éthylène est de 0,21 nm2.
| Pression de C2H4 (atmosphère) |
4,0 | 9,7 | 13,4 | 14,2 | 19,0 | 28,6 |
| Masse adsorbée (g / g de charbon) |
0,163 | 0,189 | 0,198 | 0,201 | 0,206 | 0,206 |
L’adsorption de l’azote à 90,1 K sur un certain
solide donne les volumes suivants, ramenés aux conditions TPN, adsorbés par
gramme de solide aux pressions relatives indiquées :
| P / P0 | 0,50 | 0,10 | 0,15 | 0,20 | 0,25 |
| V (cm3 ) | 51,3 | 58,8 | 64,0 | 68,9 | 74,2 |
En utilisant un graphe convenable, montrez que l’adsorption suit l’isotherme de Brunauer; Emmett et Teller.
- En déduire les valeurs de v0 et de C.
- Évaluer la surface active du solide (en m2 / g) et la valeur Ea - EL, où Ea et EL (en J·mol-1) représentent respectivement la chaleur d’adsorption et de liquéfaction de l’azote.
L’adsorption du butane sur un gramme de catalyseur à 0 ºC donne les valeurs suivantes :
| P (mmHg) | 56,39 | 89,47 | 125,22 | 156,61 | 179,30 | 187,46 |
| V (cm3, TPN) | 17,09 | 20,62 | 23,74 | 26,09 | 27,77 | 28,30 |
La grandeur P est la pression de vapeur du butane en équilibre avec le catalyseur. La surface de section d’une molécule de butane est estimée à 0,446 nm2. À partir de ces données (P0(butane) = 774,4 mm de mercure) :
a- vérifiez graphiquement que le système obéit à la loi de BET;
b- évaluez les constantes de BET (Réponse : C = 24,06);
c- calculez la surface active du catalyseur.
Freundlich a montré que, dans certaines conditions, l’adsorption d’un soluté sur un solide dispersé dans une solution est gouvernée par la loi
y = k C1/n
Dans cette équation :
y = masse de soluté adsorbé par gramme d’adsorbant;
C = concentration à l’équilibre du soluté dans le solvant;
k et n sont des constantes propres au système considéré.
Dans le cas de l’adsorption de l’acétone par du charbon, on obtient les valeurs suivantes :
| y (10-3 mol / g) | 0,208 | 0,618 | 1,075 | 1,50 | 2,08 | 2,88 |
| C (10-3 mol / litre) | 2,34 | 14,65 | 41,03 | 88,62 | 177,69 | 268,97 |
Évaluer dans ce cas les valeurs de k et de n de l’équation de Freundlich.
L’oxyde de magnésium absorbe la silice en solution dans l’eau et peut donc servir au traitement des eaux industrielles qui alimentent les bouilloires. Soit une eau contenant 26,2 ppm de silice dont on désire réduire la teneur en silice à 4,0 ppm. On a remarqué qu’avec cette eau, on trouve :
| MgO ajouté, ppm | 75 | 100 | 150 | 200 |
| SiO2 enlevé, ppm | 17,0 | 20,0 | 23,9 | 25,2 |
En utilisant l’isotherme de
Freundlich de façon appropriée, calculez les constantes de cet isotherme ainsi que la
concentration de MgO qu’il faut ajouter pour obtenir de l’eau à 4 ppm de SiO2.
Nota : 1 ppm = 1 milligramme pour 1 000 g d’eau.
Les résultats suivants ont été obtenus pour la décomposition de l'ammoniac sur un fil de tungstène à 856 °C.
|
Pression totale (mmHg) |
Temps (sec) |
|
228 |
200 |
|
250 |
400 |
|
273 |
600 |
|
318 |
1000 |
Trouver l'ordre et la constante de vitesse de la réaction. Quel type de réaction ces résultats suggèrent-ils ?
La décomposition thermique du germane gazeux en germanium et en hydrogène se produit en premier lieu par une réaction homogène du premier ordre. Cependant, une réaction hétérogène d'ordre zéro se développe graduellement et évolue parallèlement à la première, à mesure qu'un film de germanium se dépose à la surface du réacteur.
GeH4 ® Ge + 2 H2
Trouver une expression pour la vitesse de disparition du germane.
L'étude d'un système réactionnel d'un mélange de deux gaz A et B a montré qu'une réaction chimique avait lieu entre une molécule gazeuse de A et une molécule adsorbée de B.
Selon Hinshelwood la décomposition de N2O sur le platine à 741 °C est régie par la relation :
![]()
où a est la pression initiale de N2O
(95 mmHg)
x mesure la décroissance de cette pression et
k et b sont des constantes.
À partir des données suivantes, déterminez graphiquement k et b.
| temps (s) | 315 | 750 | 1 400 | 2 250 | 3 450 |
| x (mmHg) | 10 | 20 | 30 | 40 | 50 |
Calculez le temps de demi réaction à 741 °C lorsque la pression initiale est de 200 mmHg.
Delmon, B., Introduction à la cinétique hétérogène, Édition TECHNIP, Paris, 1969; QD 501 D359.
Barret, P., Cinétique hétérogène, Gauthier-Villars, Paris, 1973.
Perrin, R. et J.-P. Scharff, Chimie industrielle, 2 e édition, Masson, Paris, 1997.
Moulay, S., Bref historique des polymères, L'actualité chimique, Société française de chimie, 31-43, décembre 1999.
V. Monteil, V., La catalyse de polymérisation : repousser les limites, L'actualité chimique, Société française de chimie, 30-35, décembre 2012.
Anonyme, Un point sur "Le charbon actif", L'actualité chimique, Société chimique de France, N° 396, 63-64 (mai 2015).
Sur le NET :
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes industries, les Fondations, ...
Chercher, par exemple, avec les mots clefs : isotherme et BET.
Un lien (il y en a d'autres) relatif à LANGMUIR : http://nobelprizes.com/nobel/chemistry/1932a.html. (site visité le 2020-11-06).
Dernière mise à jour : 2021-07-02.
x x x x x x x x x x x