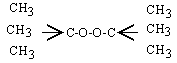CHAPITRE 10
LES RÉACTIONS RADICALAIRES
|
Objectifs Dans le concert des collisions, on n'a pas considéré le cas de systèmes mettant en jeu uniquement des entités radicalaires, celles qui ont un spin magnétique.
Le cas tout juste présenté est un cas limite de ce que l’on peut observer.
|
Objectifs spécifiques
- Comprendre la nature et les spécificités des réactions de type radical-radical.
- Être capable d’appliquer le mécanisme de RICE-HERZFELD aux réactions de décomposition d’ordre 1 ou d’ordre fractionnaire.
- Comprendre les mécanismes de polymérisation radicalaire.
- Comprendre les mécanismes d’interception de radicaux.
Ce chapitre reprend certains développements déjà réalisés en mettant l’accent sur certaines propriétés. En effet, certains systèmes réactionnels, soit qu’ils ont fait l’objet d’études diversifiées, soit qu’ils ont des propriétés particulières et de ce fait ils méritent des considérations à part. Il faut ajouter qu’il existe peu de systèmes réactionnels qui ne mettent en cause soit des réactions du type radical-radical, soit du type ion-molécule. À ce titre, ils demandent au moins un minimum d’attention.
Parce que les réactions radical-radical se réduisent au "mariage" de deux électrons célibataires par couplage de spins antiparallèles, elles n’ont pas ou peu d’énergie d’activation. Le facteur de BOLTZMANN leur est donc très favorable. Hormis quelques cas, les réactions de type radical-radical sont plus probables que les réactions radical-molécule à la condition, bien entendu, que les concentrations radicalaires soient convenables.
2. Réaction radical-radical
a- Le rapport dismutation/combinaison
Les réactions de radicaux R• + R•' ne se traduisent pas nécessairement et uniquement en une combinaison (formation du produit R¾R'). En effet, la formation de la nouvelle liaison, en d’autres termes le couplage magnétique des deux électrons célibataires est en général très exothermique. La molécule nouvellement formée est très énergétique. Elle a la possibilité de se décomposer en un alcène et un alcane. Le résultat net est un transfert d’un atome d’hydrogène d’un radical vers l’autre : c’est une réaction de dismutation :
Par exemple :
| C2H5• + C2H5• | ® n-C4H10 | combinaison |
| C2H5• + C2H5• | ® C2H4 + C2H6 | dismutation |
Dans le cas des radicaux hydrocarbonés et saturés, en phase gazeuse, les proportions des réactions de dismutation sont relativement faibles (T = 298 K) :
|
Réaction de combinaison |
probabilité | |
| C2H5• + n-C3H7• | ® n-C5H12 | 1,00 |
| Réaction de dismutation | ||
| C2H5• + n-C3H7• | ® C2H4 + C3H8 | 0,057 |
| C2H5• + n-C3H7• | ® C2H6 + C3H6 | 0,065 |
Dans le cas des radicaux méthyles, la formation du radical méthylène, :CH2, est très endothermique. Il en résulte que la dismutation des radicaux méthyles ne conduit qu’à la formation du méthane :
| CH3• + iso-C3H7• | ® iso-C4H10 | 1,00 |
| CH3• + iso-C3H7• | ® CH4 + C3H6 | 0,163 |
Le rapport dismutation/combinaison dans le cas de réactions mettant en jeu des radicaux insaturés, sont moins bien connus. Cependant les réactions de dismutation semblent demeurer de peu d’importance.
|
Réaction de combinaison |
probabilité | |
| C2H3• + C2H5• | ® CH3-CH2-CH=CH2 | 1,00 |
| Réaction de dismutation | ||
| C2H3• + C2H5• | ® C2H4 + C2H4 | 0,03 |
| C2H3• + C2H5• | ® C2H2 + C2H6 | 0,12 |
Ces données numériques supportent, en première approximation, qu’on puisse ignorer les réactions de dismutation, bien que, pour des résultats précis, on doive en tenir compte. Les radicaux tertiaires font exception à cette règle : ils dismutent très facilement [Terry, J. O. et J. H. Futrell, Can. J. Chem., 46, 664-665 (1968)] :
|
Réaction de combinaison |
probabilité | |
| t-C4H9• + C2H5• | ® isohexane | 1,00 |
| Réaction de dismutation | ||
| t-C4H9• + C2H5• | ® iso-C4H8 + C2H6 | 0,50 |
| t-C4H9• + C2H5• | ® iso-C4H10 + C2H4 | 0,28 |
b- L’énergie d’activation
Comme on vient de le mentionner plus haut, les réactions du type R• + R• consiste essentiellement en un couplage de deux spins électroniques. L’énergie d’activation est essentiellement zéro. Les constantes de vitesse de ce type de réaction sont donc pratiquement égales entre elles (Tableau 10.1) et de plus égale à la constante de vitesse de la collision physique. Il faut cependant introduire un facteur géométrique 1/2 qui tient compte de l’orientation réciproque des spins électroniques : ils doivent être antiparallèles. De plus, si le radical est volumineux, les deux champs magnétiques peuvent ne pas se voir et la collision est inefficace. Deux radicaux n-octyles doivent se percuter de façon appropriée, c'est-à-dire par leur extrémité radicalaire ...
Tableau 10.1. Exemples de vitesses de réactions radicalaires
| Radicaux (•R1, •R2) | 10-12 k (cm3.mol-1.s-1) |
| •CH3, •CH3 | 19,0 |
| •CH3, •C2H5 | 27,2 |
| •CH3, iso-C3H7• | 22,0 |
| •CH3, tert-C4H9• | 12,5 |
| •C2H5, •C2H5 | 8,77 |
| iso-C3H7•, iso-C3H7• | 5,96 |
| tert-C4H9•, tert-C4H9• | 4,31 |
| Arthur, N. L. et C. Anastasi, Bull. Soc. Chim. Belg., 92(6-7), 647 (1983). | |
On conçoit donc que dans un milieu réactionnel, la formation de produits issus de l’interaction entre des radicaux A•, B•, C•,... est directement régie par la probabilité de collisions entre les paires de radicaux. Soit un milieu réactionnel dans lequel on trouve le radicaux A• et B• :
[a] A• + A• ® produits, vitesse va
[b] B• + B• ® produits, vitesse vb
[c] A• + B• ® produits, vitesse vc
Soit à calculer le rapport des vitesses des réactions croisées, Â
![]()
La grandeur vi du membre de droite de cette équation représente la vitesse de la réaction i. L’estimation des vitesses revient à calculer la probabilité de tirer dans une boîte les radicaux appropriés. Ainsi, la probabilité de réussir la réaction [a] est égale à la probabilité de sortir deux fois de suite le radical A• (a est une constante de proportionnalité) :
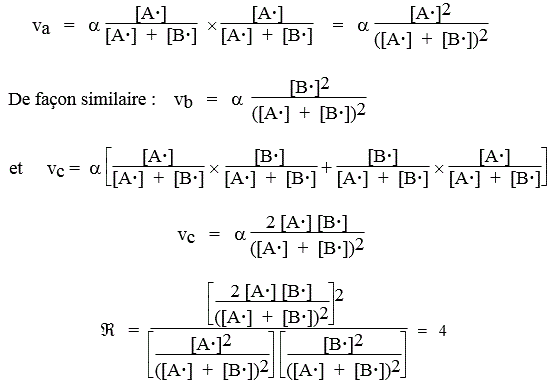
et :
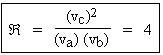
En fait le rapport  doit être un peu plus grand que 4. La vitesse d’une réaction chimique ne mettant pas en jeu d’énergie d’activation est de la forme :
vAB = a dAB2 µAB-1/2
dAB est le diamètre de collision et
µAB est la masse réduite des partenaires en collision.
Dans ce cas  devient Â' (Voir équation 2.5 et Tableau 10.2) :
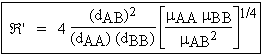
Tableau 10.2. Exemples de valeurs obtenues pour Â'
| Radicaux (R1, R2) | Valeurs théoriques | Valeurs expérimentales |
| •CH3, •C2H5 | 4,24 | 4,00 |
| •CH3, iso-C3H7• | 4,75 | 4,00 |
| •CH3, tert-C4H9• | 5,29 | 1,69 |
| •CH3, •C3H5 | 4,66 | 2,56 |
| •CCl3, •CH3 | 6,70 | 0,18 |
| Garland, L. J. et K. D. Kyle, J. Phys. Chem., 94, 4941 (1990). | ||
Il existe cependant deux cas types où les radicaux ne réagissent pas entre eux. Le premier cas survient lorsque les concentrations radicalaires sont extrêmement faibles. Les radicaux ont alors la possibilité d’avoir un temps de vie suffisamment long pour diffuser jusque sur les parois du réacteur. En alternative, et dans les cas où l’énergie d’activation, EA, des réactions du type radical-molécule n’est pas très élevée, cette dernière possibilité offre une voie de consommation des radicaux. C’est particulièrement le cas de l’addition des atomes d’hydrogène sur les doubles liaisons. Même en présence de concentrations importantes de radicaux, la réaction d’addition ne peut être ignorée :
•H + CH3CH=CH2 ® s-C3H7•
Cette réaction transforme un atome d’hydrogène en un radical alkyle. Le système se retrouve alors dans la situation générale évoquée plus haut et ceci d’autant plus que la formation d’une molécule d’hydrogène (via la réaction •H + •H) est soumise à la réversibilité : voir paragraphe 2.10.
3. Le mécanisme de RICE-HERZFELD
Dans le cas de décomposition de matière organique par voie thermique, le mécanisme de nature radicalaire, le mécanisme de RICE-HERZFELD, est schématisé de la façon suivante. C’est un mécanisme en chaîne :
| a- amorçage (initiation) : | ||||
| [1] M | ® | •R1 + •R1' | k1 | |
| b- propagation : | ||||
| [2] •R1 + M | ® | •R2 + R1H | k2 | |
| [3] •R2 | ® | •R1 + M' | k3 | |
| c- rupture (terminaison) : | ||||
| [4] •R1 + •R1 | ® ... | Produits de combinaison
et de dismutation |
k4 | |
| [5] •R1 + •R2 | ® ... | k5 | ||
| [6] •R2 + •R2 | ® ... | k6 |
Le radical •R1 propage la chaîne dans une réaction bimoléculaire: c’est un porteur de chaîne aussi appelé centre actif. Rappelons aussi que les réactions [4] à [6] ne commandent pratiquement aucune énergie d’activation. La réaction globale est la somme des réactions comprises dans la période de propagation de chaîne. En effet, les périodes d’amorçage et de rupture de chaîne n’ont lieu qu’un nombre de fois limité devant le nombre de fois que se répète le cycle de la propagation. Donc, la réaction globale est :
[7] M ® R1H + M'
2-a- Cinétique globale d’ordre 3/2
Supposons que la concentration des radicaux •R1 soit beaucoup plus grande que celle du radical •R2.
[•R1] >> [•R2 ]
| a- amorçage (initiation) : | ||||
| [1] M | ® | •R1 + •R1' | k1 | |
| b- propagation : | ||||
| [2] •R1 + M | ® | •R2 + R1H | k2 | |
| [3] •R2 | ® | •R1 + M' | k3 | |
| c- rupture (terminaison) : | ||||
| [4] •R1 + •R1 | ® ••• | Produits de combinaison et de dismutation |
k4 |
Compte tenu de ce qui a été dit plus haut à propos des réactions radical-radical, la réaction [4] est beaucoup plus probable que les réactions [5] et [6]. Appliquons maintenant le principe de quasi-stationnarité aux radicaux •R1 et •R2 :
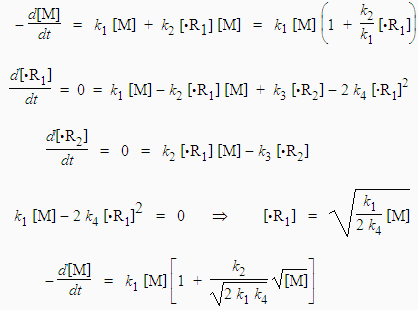
Comme la réaction suit un mécanisme en chaîne, la réaction d’amorçage est négligeable devant les réactions de propagation de chaîne. Par conséquent,
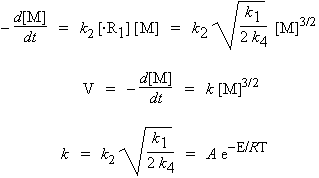
En identifiant les facteurs pré-exponentiels et les énergies d’activation :
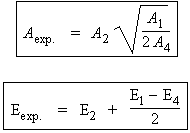
On montre que généralement, l’énergie d’activation mesurée, Eexp, est de l’ordre de 210 kJ·mol-1 au lieu de 335 normalement prévus pour l’énergie d’activation de la réaction d’amorçage.
La longueur de chaîne est
le nombre de fois que se produit la boucle de propagation par rapport au nombre de fois
qu’a lieu l’étape d’amorçage. En d’autres termes la
longueur de
chaîne,
![]() , est égal à la vitesse de formation de M' divisée par la
vitesse de la réaction d’amorçage.
, est égal à la vitesse de formation de M' divisée par la
vitesse de la réaction d’amorçage.
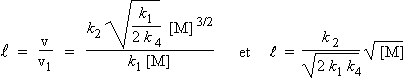
Soit le mécanisme de décomposition de l’acétaldéhyde :
| [1] | CH3CHO | ® •HCO + •CH3 | k1, | E1 = 317,7 kJ.mol-1 |
| [2] | •CH3 + CH3CHO | ® •CH2CHO | k2, | E2 = 41,8 kJ.mol-1 |
| [3] | •CH2CHO | ® •CH3 + CO | k3, | E3 = 75,2 kJ.mol-1 |
| [4] | •CH3 + •CH3 | ® C2H6 | k4, | E4 = 0 kJ.mol-1 |
On ajoute que l’on ignore ce que devient le radical •HCO, et que la réaction [4] est une réaction thermoléculaire, et donc se fait à haute pression. La résolution de ce système conduit à :
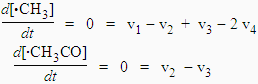
Þ v1 = 2 v4, Þ k1 [CH3CHO] = 2 k4 [•CH3]2
La vitesse de la réaction est telle que :
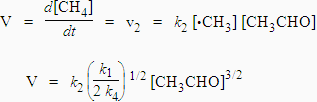
L’énergie d’activation expérimentale est : Eexp = E2 + (E1 - E4 )/2 = 200,6 kJ.mol-1
La longueur de chaîne,
![]() , est telle que :
, est telle que :
![]()
2-b- Cinétique globale d’ordre 1/2
Dans le cas où la concentration des radicaux •R2 est beaucoup plus grande que celle des radicaux •R1,
[•R1] << [•R2 ]
| a- amorçage (initiation) : | ||||
| [1] M | ® | •R1 + •R1' | k1 | |
| b- propagation : | ||||
| [2] •R1 + M | ® | •R2 + R1H | k2 | |
| [3] •R2 | ® | •R1 + M' | k3 | |
| c- rupture (terminaison) : | ||||
| [6] •R2 + •R2 | ® ... | Produits de combinaison et de dismutation |
k6 |
la réaction de rupture de chaîne doit être identifiée à la réaction [6]. En appliquant le principe de quasi-stationnarité à ces deux radicaux et en négligeant les étapes d’amorçage et de rupture devant l’étape de propagation, on obtient :
et |
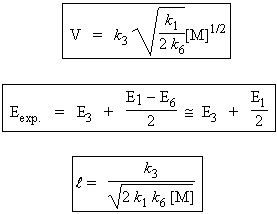 |
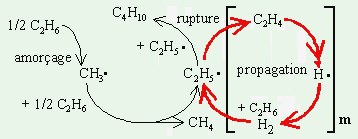
Soit la décomposition de l’éthane selon le mécanisme suivant. La réaction d’amorçage est une réaction d’ordre 1.
| [1] | CH3-CH3 | ® •CH3 + •CH3 | k1 |
| [2] | •CH3 + C2H6 | ® •C2H5 + CH4 | k2 |
| [3] | •C2H5 | ® •H + C2H4 | k3 |
| [4] | •H + C2H6 | ® H2 + •C2H5 | k4 |
| [5] | 2 C2H5• | ® n-C4H10 | k5 |
À haut taux de conversion, la réaction d’addition de l’atome d’hydrogène sur l’éthylène formé vient ajouter une autre boucle réactionnelle.
|
|
Figure 10.1. Schéma réactionnel de décomposition
thermique de l’éthane.
Résolution :
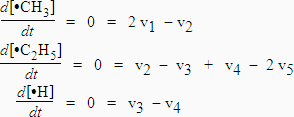
La
première équation entraîne que :
Þ
2 k1
[C2H6]
=
k2
[•CH3]
[C2H6]
Þ
[•CH3]
=
2 k1
/k2
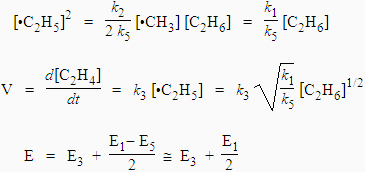 |
|
| Or, | |
| L’énergie d’activation est : |
Quant à la longueur de chaîne,
![]() , elle est telle que :
, elle est telle que :
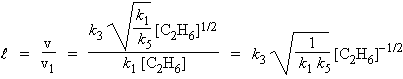
2-c- Cinétique globale d’ordre 1
Si la réaction de terminaison est plutôt la réaction [5], [•R1] ~ [•R2 ], on montre que la réaction globale est d’ordre 1:
| a- amorçage (initiation) : | ||||
| [1] M | ® | •R1 + •R1' | k1 | |
| b- propagation : | ||||
| [2] •R1 + M | ® | •R2 + R1H | k2 | |
| [3] •R2 | ® | •R1 + M1' | k3 | |
| c- rupture (terminaison) : | ||||
| [5] •R1 + •R2 | ® ••• | Produits de combinaison et de dismutation |
k5 |
et |
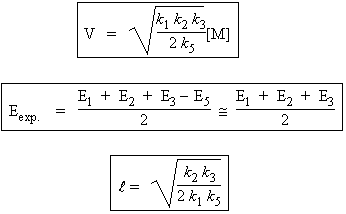 |
Soit la pyrolyse du 1,2-dichloroéthane vers 362-485 °C. Elle procède selon un mécanisme en chaîne amorcée en une réaction d’ordre 1 :
| [1] | ClCH2-CH2Cl | ® •ClCH-CH2Cl + •CCl | k1 |
| [2] | •Cl + ClCH2-CH2Cl | ® •ClCH-CH2Cl + HCl | k2 |
| [3] | •ClCH-CH2Cl | ® •Cl + ClCH=CH2 | k3 |
| [4a] | •Cl + •ClCH-CH2Cl | ® HCl + ClCH=CH2 | k4a |
| [4b] | ® ou Cl2CH-CH2Cl | k4b |
La réaction globale est égale à la somme des étapes de propagation de chaîne : v2 + v3 :
[5] ClCH2-CH2Cl ® ClCH=CH2 + HCl
L’application du principe de quasi-stationnarité donne :
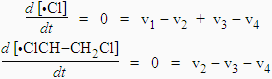
En additionnant : Þ 0 = v1 - 2 v4
Þ k1 [ClCH2-CH2Cl] = 2 k4 [•Cl] [•ClCH-CH2Cl]
et k2 [•Cl] [ClCH2-CH2Cl] = k3 [•ClCH-CH2Cl]
[•ClCH-CH2Cl] = (k2 /k3) [•Cl] [ClCH2-CH2Cl]
k1 [ClCH2-CH2Cl] = 2 k4 [•Cl] (k2 /k3) [•Cl] [ClCH2-CH2Cl]
et |
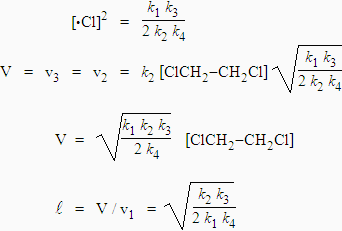 |
|
La longueur de chaîne est : |
On donne les valeurs des
constantes suivantes (les énergies sont exprimées en cal.mol-1) :
k1 = 1013 e-77 900/RT s-1
k2 = 1010,8 e-3 100/RT L.mol-1.s-1
k3 = 1013,8 e-23 000/RT s-1 et
k4 = 1011 L.mol-1.s-1
On calcule les valeurs : [•Cl] = 2,14 10-13
mol.L-1
et [•ClCH-CH2Cl] = 61 10-13
mol.L-1
On peut donc raisonnablement
éliminer la réaction de recombinaison de deux atomes de chlore, d’autant qu’il faudrait
tenir compte d’une réaction thermoléculaire sous pression élevée. Par contre on
devrait plutôt tenir compte de la réaction de recombinaison de deux radicaux •ClCH¾CH2Cl
dont les produits sont, entre autres produits de dismutation, le 1,2,3,4-tétrachlorobutane.
D’autres études ont aussi montré que les réactions de terminaison ont aussi
lieu sur les parois du réacteur. La longueur de chaîne,
![]() , à 700 K indique que celle-ci
est de l’ordre de 2,16 108. Les atomes de chlore subissent 2,16 108 collisions avant la
rupture. Le calcul du libre parcours moyen peut être effectué (voir
Chapitre
2) :
, à 700 K indique que celle-ci
est de l’ordre de 2,16 108. Les atomes de chlore subissent 2,16 108 collisions avant la
rupture. Le calcul du libre parcours moyen peut être effectué (voir
Chapitre
2) :
l = 12,6 10-8 m
Par conséquent, entre la
naissance de la chaîne et la rupture, les atomes de chlore ont la possibilité
d’effectuer un parcours de ![]() l, ou encore de 27,2 mètres! Ce parcours
n’est évidemment pas en ligne droite,
mais plutôt de façon désordonnée (mouvement brownien). À moins d’avoir un réacteur
de capacité importante, on peut s’attendre à une terminaison au moins partielle de la
chaîne sur les parois.
l, ou encore de 27,2 mètres! Ce parcours
n’est évidemment pas en ligne droite,
mais plutôt de façon désordonnée (mouvement brownien). À moins d’avoir un réacteur
de capacité importante, on peut s’attendre à une terminaison au moins partielle de la
chaîne sur les parois.
Autre exemple : soit la décomposition de l’éther diméthylique :
| [1] | CH3OCH3 | ® •CH3 + •OCH3 | k1, E1 = 336 kJ.mol-1 |
| [2] | •CH3 + CH3OCH3 | ® •CH2OCH3 + CH4 | k2, E2 = 62,7 kJ.mol-1 |
| [3] | •CH2OCH3 | ® •CH3 + HCHO | k3, E3 = 158,8 kJ.mol-1 |
| [4] | •CH3 + •CH2OCH3 | ® CH3CH2OCH3 | k4, E4 = 33,6 kJ.mol-1 |
Selon le mécanisme de RICE-HERZFELD :
|
v = |
E = (1/2) (E1 + E2 + E3 - E4) = 261,2 kJ.mol-1
Les radicaux •OCH3 se décomposent en •H + HCHO. Les atomes d’hydrogène amorcent une autre chaîne ce qui, globalement, fait baisser l’énergie d’activation expérimentale à 244,5 kJ.mol-1.
4. Le peroxyde de tertiobutyle
Le peroxyde de tert-butyle est un composé formé d’une liaison centrale O-O relativement faible. C’est donc un produit qui, même à la température ordinaire, tend à se décomposer faiblement. Il génère ainsi des radicaux libres en concentrations suffisantes de telles sortes que, si ce composé est ajouté à un composé susceptible de polymérisation radicalaire, il en amorce la polymérisation.
| Sa structure est : |
|
Les réactions globales sont :
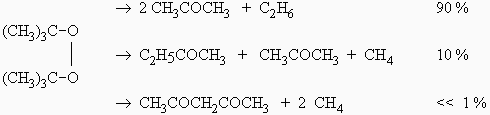
et le mécanisme est :
| [1] | M | ® 2 (CH3)3CO• | k1 |
| [2] | (CH3)3CO• | ® •CH3 + CH3COCH3 | k2 |
| [3] | •CH3 + CH3COCH3 | ® •CH2COCH3 + CH4 | k3 |
| [4] | •CH3 + •CH2COCH3 | ® C2H5COCH3 | k4 |
| [5] | •CH3 + •CH3 | ® C2H6 | k5 |
Le radical méthyle ne réagit pas avec le peroxyde. La décomposition ne suit donc pas le mécanisme de RICE-HERZFELD. La résolution du système utilise les mêmes principes.
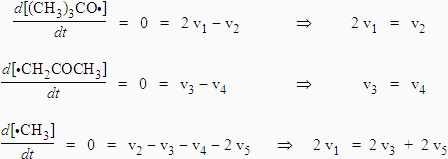
Au temps t = 0, il n’y a pas ou très peu d’acétone, on peut donc négliger v3 devant v5. On obtient donc :
|
k1 [M] = k5 [•CH3]2 Þ [•CH3] = |
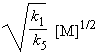 |
Si la pression du peroxyde est de 7 mmHg, à 300 °C on obtient une concentration de radicaux méthyles de 0,017 mol.litre-1, donc une concentration relativement importante pour une espèce réactive.
5. La polymérisation radicalaire
Certains systèmes sont connus pour leur mécanisme de polymérisation radicalaire. Soit par exemple,
| a- Amorçage | |||
|
[a] |
I | ® •R1 | ka |
| b- Propagation | |||
| [1] | •R1 + M | ® •R2 | k1 |
| [2] | •R1 + M | ® •R3 | k2 |
|
--------------------------------------------------- etc. |
|||
| [n -1] | •Rn-1 + M | ® •Rn | kn-1 |
| [n] | •Rn + M | ® •Rn+1 | kn, ... etc. |
| c- Terminaison | |||
|
[i,j] |
•Ri + •Rj | ® •Ri,,j | ki,j, ... etc. |
La chaîne cinétique de réactions forme une chaîne polymérique. La vitesse de la réaction d’amorçage est telle que :
Vam. = ka [I]
d[•R1] / dt = ka [I] - k1 [•R1] [M] - k1,n Sn[•R1] = 0
d[•R2] / dt = k1 [•R1] [M] - k2 [•R2] [M] - k2,n Sn[•R2] = 0
d[•R2] / dt = kn-1 [•Rn-1] [M] - kn [•Rn] [M] - kn,n Sn[•Rn] = 0
ka [I] = k1 [•R1] [M] + k1,n Sn[•R1]
k1 [•R1] [M] = k2 [•R2] [M] + k2,n Sn[•R2]
k2 [•R2] [M] = k3 [•R3] [M] + k3,n Sn[•R3]
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
kn-1 [•Rn-1] [M] = kn [•Rn] [M] + kn,n Sn[•Rn]
En additionnant membre à membre,
ka [I] = ( k1 [•R1] + k2 [•R2] + k3 [•R3] + . . . + kn,n Sn[•Rn] ) [M]
Mais k1,n = k2,n = k3,n = ... = kn,n puisque ce sont des réactions identiques de condensation d’un radical sur le monomère M. Donc :
Vam = ki,j Sn[•Rn] Sn[•Rn] et
![]()
La vitesse de disparition du monomère est :
- k[M] / dt = k1 [•R1] [M] + k2 [•R2] [M] + k3 [•R3] [M] + ... + kn [•Rn] [M]
- d[M] / dt = ki [M] Sn[•Rn].
En effet, toutes les constantes k1, k2, k3, ki ...représentent des réactions de condensation identiques.
| Þ |
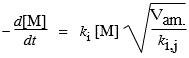 |
La vitesse de disparition du monomère dépend donc de la vitesse d’amorçage.
M + M ® •R + M + X; Vam. = ktherm. [M]2
| et |
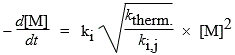 |
M + C ® •R + C + X; Vam. = kcat. [M] [C] et
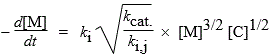
M + hn ® •R + C; Vam. = Ia f et
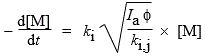
Le mécanisme de polymérisation radicalaire tout juste présenté se termine lorsque deux chaînes hydrocarbonées en croissance se recombinent. Il existe au moins un autre mécanisme de rupture de chaîne. Celle-ci se fait par transfert de chaîne cinétique vers le monomère :
•Ri + M ® RiH +
•R1;
ki
Il n’est pas toujours facile d’identifier la nature radicalaire d’un mécanisme réactionnel, et encore moins facile d’en identifier les intermédiaires radicalaires. On dispose cependant de tests qui permettent d’obtenir des informations pertinentes. L’usage d’intercepteurs radicalaires est d’un emploi facile. Les radicaux disposent d’un électron non apparié et l’on a vu que leur principale réaction de disparition est par "re-mariage" de spin électronique. Cela suggère l’addition de composés paramagnétiques comme l’oxygène moléculaire ou l’oxyde nitrique en phase gazeuse. Les radicaux vont en effet réagir très rapidement avec l’un ou l’autre de ces deux additifs.
•Ri + O2 ® Ri¾O¾O•
Bien souvent, le nouveau radical Ri¾O¾O• n’a pas les mêmes propriétés que le radical •Ri : les réactions entre les radicaux hydrocarbonés ne se combinent plus pour donner des hydrocarbures, mais donnent plutôt des hydrocarbures oxygénés. Les polymérisations radicalaires sont très sensibles à la présence de traces d’oxygène. On a vu, également, que l’oxygène peut provoquer d’autres réactions (voir Chapitre 5). Par conséquent, l’effet de l’oxygène, ajouté à un milieu réactionnel, ne constitue pas en lui-même une preuve irréfutable de la nature radicalaire d’un mécanisme réactionnel. Il ajoute cependant au faisceau de preuves.
Il existe des intercepteurs plus révélateurs non seulement de la nature radicalaire du mécanisme, mais aussi de l’identification des radicaux libres intermédiaires. Considérons l’usage de l’iodure d’hydrogène, HI. Ce composé a une liaison H-I relativement faible. Il s’ensuit qu’un radical pourra arracher son atome d’hydrogène dans une réaction ayant une faible énergie d’activation :
CH3• +
HI ® CH4 +
I• Ea = 9,6 kJ·mol-1
L’augmentation du rendement en méthane lorsque l'on ajoute quelques pour cents d'iodure d’hydrogène permet de mesurer les rendements de nombreux radicaux. La technique peut être sophistiquée quelque peu. En substituant HI par son frère deutérié on peut avoir accès à la structure du radical intermédiaire en ajoutant une analyse, par spectrométrie de masse, du produit monodeutérié RD formé :
CH3CH(•)CH3
+ DI ®
CH3CHDCH3 + I•
CH3CH2CH2(•) + DI
®
CH3CH2CH2D + I•
D’autres intercepteurs ont été utilisés : le bromure d’hydrogène, HBr, l’hydrogène sulfureux, H2S, de façon similaire à l’iodure d’hydrogène, HI. Il faut noter que le radical méthylène réagit en présence de DI pour former le méthane-d2. Il est donc possible de distinguer entre les radicaux méthyles et méthylènes. Les alcènes ont aussi été utilisés dans des milieux saturés,...
7. Conclusions
|
1- À partir du tableau 10.1, calculer le rapport des vitesses de réactions croisées  dans le cas des couples de radicaux •CH3 et •C2H5, •CH3 et iso-C3H7•, •CH3 et tert-C4H9• et comparer vos résultats avec ceux apparaissant dans le tableau 10.2.
2- La décomposition thermique de l’acétaldéhyde a été étudiée dans des conditions particulières [Letort, M., J. Chim. Phys., 34, 206 (1937)] :
[1] M + CH3CHO ® •CH3 + •CHO + M k1
[2] •CH3 + CH3CHO ® CH4 + CH3CO• k2
[3] CH3CO• ® •CH3 + CO k3
[4] M + 2 •CH3 ® M + C2H6 k4
M représentant une molécule quelconque, évaluer, en fonction des constantes et de la concentration en acétaldéhyde, la concentration en radicaux méthyles, la vitesse de réaction et la longueur de chaîne.
3- La décomposition thermique de l’éthane a été étudiée vers 550-650 °C et sous une pression de 0,5 atm. par Küchler et al. [Z. Phys. Chem., B42, 359 (1939)]. Ces auteurs ont proposé le mécanisme suivant :
[1] C2H6 + C2H6 ® 2 •CH3 + C2H6 k1
([1'] C2H6 + C2H6 ® C2H6* + C2H6
([1''] C2H6* ® 2 •CH3 )
[2] •CH3 + C2H6 ® CH4 + •C2H5 k2
[3] •C2H5 ® C2H4 + H• k3
[4] C2H6 + H• ® H2 + •C2H5 k4
[5] •C2H5 + C2H5• ® n-C4H10 k5
En utilisant le principe de quasi-stationnarité, établissez en fonction des paramètres usuels, la réaction globale, les concentrations [•H] et [•C2H5], la vitesse de la réaction globale, la longueur de chaîne et l’énergie d’activation expérimentale à la pression atmosphérique.
À partir des données numériques apparaissant dans le tableau suivant, établissez les valeurs numériques de [•H], [•C2H5] et de la longueur de chaîne.
| Réaction : | Facteur pré exponentiel | Énergie d’activation |
| [1] | 6,5 1017 cm3.mol-1.s-1 | 70,2 kcal.mol-1 |
| [2] | 2,0 1011 cm3.mol-1.s-1 | 10,4 kcal.mol-1 |
| [3] | 3,0 1014 s-1 | 39,5 kcal.mol-1 |
| [4] | 3,4 1012 cm3.mol-1.s-1 | 6,8 kcal.mol-1 |
| [5] | 7,6 1012 cm3.mol-1.s-1 | 0,0 kcal.mol-1 |
4- Afin de décider quelle réaction de terminaison est efficace dans la pyrolyse de l’éthane, quelle(s) méthode(s) d’analyse (laboratoire) proposeriez-vous pour choisir entre les réactions [5] et [6] ?
[1] C2H6* ® 2 •CH3
[2] •CH3 + C2H6 ® CH4 + •C2H5
[3] •C2H5 ® C2H4 + H•
[4] •H + C2H6 ® H2 + •C2H5
[5] •C2H5 + •C2H5 ® n- C4H10
[6] •C2H5 + •CH3 ® C3H8
5- Soit la réaction : CH3• + HCl ® CH4 + •Cl
La constante de vitesse, k (10 cm3•molécule-1•s-1), de cette réaction a été mesurée à diverses températures [Russell, J. J. et al., Int. J. Chem. Kinetics, 20, 759-73 (1988)]. Calculez l’énergie d’activation de la réaction et son facteur pré exponentiel.
| T (K) | 296 | 316 | 342 | 372 | 406 | 450 | 495 |
| k | 5,2 | 5,5 | 6,6 | 7,8 | 9,0 | 10,4 | 11,6 |
6- Dans l’étude de la photochimie de l’éthane, il se forme des atomes d’hydrogène et des radicaux méthyles. Pour mesurer ces intermédiaires radicalaires, on ajoute un peu de propène. En supposant que cet additif a une concentration très faible et qu’il ne participe pas à la photochimie, mais qu’il sert seulement à intercepter les atomes d’hydrogène, proposez l’ensemble des réactions radicalaires qui ont lieu dans ce système.
7- Dans l’étude de la photochimie du propène, les deux réactions suivantes sont à peu près d’égale importance. Écrivez toutes les réactions radicalaires qui en découlent (négliger les réactions de type radical hydrocarboné + propène) :
CH3-CH=CH2 + hn ® H• + •CH2-CH=CH2 et
CH3-CH=CH2 + hn ® CH3• + •CH=CH2
Trotman-Dickenson, A. F. et G. S. Milne, Tables of Bimolecular Gas Reactions, N.S.R.D.S.- N.B.S. 9, 1967.
Ratajezak, E. et A. F. Trotman-Dickenson, Suplementary Tables of Bimolecular Gas Reactions, OSTI, Univ. of Wales Inst. of Science and Technology, 1970.
Kerr, J. A. et M. J. Parsonage, Evaluated Kinetic Data on Gas Phase Addition Reactions, CRC Press, Cleveland, Ohio, 1972; QD501 K41.
Sur le NET :
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes industries, les Fondations, ...
On y trouvera une quantité importante d'informations relatives à des réactions du type atome + molécule diatomique et de beaucoup d'autres types (y inclus les DH de réaction). Exemple : https://webbook.nist.gov/ (site visité le 2020-11-04).
Dernière mise à jour : 2021-07-02.
x x x x x x x x x x x x x