| 1.1 |
CHAPITRE 1
CARACTÈRES GÉNÉRAUX DE LA CINÉTIQUE DES RÉACTIONS HOMOGÈNES
|
La cinétique chimique veut exprimer la relation qui existe entre la réaction chimique et le temps. Pourquoi certaines réactions sont infiniment lentes alors que la thermodynamique affirme par ailleurs qu’elles sont réalisables? Pourquoi certaines réactions très lentes deviennent soudainement très rapides, voire explosives? Avant d’entrer dans le cœur du sujet, il est utile de
|
Objectifs spécifiques
- Assimiler le vocabulaire utilisé dans le domaine de la cinétique chimique.
- Comprendre les différentes définitions de vitesse de réaction.
- Comprendre les différences entre réaction élémentaire et réaction globale, entre ordre et molécularité, entre enthalpie et énergie d’activation de réaction.
- Aborder les relations qui existent entre la vitesse d’une réaction et le mécanisme réactionnel, entre la vitesse de réaction et la température de réaction, entre la thermochimie et la cinétique chimique.
1. Notions de mécanismes élémentaires
Mécanisme élémentaire, molécularité
Une réaction chimique peut-être le résultat d’une réaction élémentaire qui se fait en une seule étape. Cependant, le plus souvent c’est le résultat d’une suite de réactions élémentaires. Soit une réaction élémentaire :
A + B ® M + N
Cela signifie (voir ultérieurement) que la rencontre des deux particules A et B produit en une seule étape les espèces M et N. Dans ce cas, on dira que la molécularité de la réaction est le nombre de particules (atomes, molécules, radicaux, ions positifs ou négatifs,...) nécessaires pour la réalisation de la réaction. C’est un nombre positif différent de zéro qui ne peut être déterminé que par la connaissance du mécanisme de réaction. En fait ce genre, de réaction où en une étape on passe directement des réactifs aux produits stables, est extrêmement rare. L’exemple universellement cité est la réaction :
H2 + I2 ® 2 HI (molécularité = 2)
Il est difficile de démontrer que cette réaction est bien le résultat d’une seule étape réactionnelle. Il est en fait plus fréquent de démontrer qu’une réaction se réalise à travers une série de réactions élémentaires consécutives dont la somme devient une réaction globale. La molécularité n’a alors plus de signification. On verra d’ailleurs plus loin qu’il n’est pas certain que la réaction précédente se réalise directement dans une étape élémentaire.
D’autres exemples :
cyclo-C4H8 ® 2 C2H4
CO + NO2 ® CO2 + NO·
Cette réaction a bien lieu en une étape lorsque la température est supérieure à 600 K. Cependant, à une température inférieure à 500 K, elle procès à travers les deux étapes élémentaires suivantes :
NO2+ NO2 ® NO3+ NO•
CO + NO3 ® CO2 + NO2
Soit la réaction :
2 NO• + 2 H2 ® 2 H2O + N2
Cette réaction, si elle était élémentaire impliquerait que quatre molécules (les réactifs) se rencontrent en même temps pour donner lieu à une réaction qui, simultanément réduit l’azote et oxyde l’hydrogène. Il devrait y avoir transfert simultané de quatre électrons. Cela est peu probable. On préfère plutôt un mécanisme un peu plus compliqué, mais qui réalise, à chaque étape un minimum de transformation.
Ce qu’en d’autres termes on pourrait appeler, une loi du travail minimum. Le mécanisme suivant répond à ce souci de simplification (la somme des deux réactions élémentaires consécutives correspond bien à la réaction globale) :
2 NO• + H2
®
H2O2 + N2
H2O2 + H2 ® 2 H2O
Ces notions se clarifieront plus tard. Les notions de réaction élémentaire et de molécularité seront cependant d’une grande utilité.
2. Vitesses de réaction
Il est évident qu’une réaction prend un certain temps pour se réaliser. Les vitesses extrêmes peuvent se résumer provisoirement aux explosions (vitesses très rapides) et aux phénomènes liés à l’évolution des espèces (vitesses infiniment lentes). Avant de décrire quelques définitions de vitesse, on aura besoin de préciser certaines notions de temps.
a) Définitions de temps
1- temps initial ou temps zéro, t0
C’est le moment du départ, du démarrage d’une réaction chimique. S’il est facile théoriquement de préciser cet instant, on verra qu’expérimentalement, il n’est pas toujours aisé de le déterminer.
2- temps de demi-réaction, t1/2
On verra ultérieurement une définition précise de cette notion. Disons pour le moment qu’il s’agit du temps nécessaire pour que la moitié d’une réaction soit complétée.
3- temps infini, t¥
Le temps infini par définition est une notion abstraite puisque, en particulier en cinétique chimique, personne ne peut prétendre être capable d’observer une réaction après une telle durée. En pratique, on considère que la réaction a atteint un temps infini lorsque 99,9 % des réactifs auront été transformés.
b- Définitions
Soit la réaction faisant intervenir a moles de A, b moles de B, etc. pour produire m moles de M, n moles de N, etc.
a A + b B ® m M + n N . . .
On peut exprimer la vitesse par rapport à la disparition d’un réactif ou tout aussi bien par rapport à l’apparition d’un produit. On définira la vitesse totale de la réaction globale comme étant le nombre de molécules de l’un ou l’autre des réactifs qui disparaissent ou de produits qui apparaissent par unité de temps :
| 1.1 |
On préfère, pour des fins de comparaison, parler de vitesse totale par unité de volume. On définit ainsi la vitesse spécifique :
| 1.2 |
|
Dans ces quatre égalités V est le volume du réacteur.
On utilise aussi les expressions vitesse de transformation et vitesse de disparition. Prenons un exemple pour préciser le sens de ces expressions. Soit la réaction de synthèse de l’ammoniac en phase gazeuse :
N2 + 3 H2 ® 2 NH3
Telle qu’écrite on dira que la vitesse de formation de l’ammoniac est égale au nombre de molécules d’ammoniac qui apparaissent pendant l’unité de temps ou encore que la vitesse de disparition de l’hydrogène est égale au nombre de molécules d’hydrogène qui disparaissent pendant l’unité de temps.
![]()
D’après la stœchiométrie de la réaction, il apparaît que la vitesse de disparition de l’hydrogène est trois plus grande que celle relative à l’azote. La vitesse d’apparition de l’ammoniac est 2 fois plus grande que celle de disparition de l’azote.
VNH3 = - 3 VH2 = - 2 VN2
On introduit un signe négatif devant les vitesses de disparition de l’hydrogène et d’azote pour tenir compte du fait que si le nombre de molécules d’ammoniac augmente avec le temps, ceux relatifs à l’hydrogène et à l’azote diminuent. La vitesse, V, de la réaction telle que précédemment écrite correspond à la vitesse de disparition de l’azote ou à la vitesse de formation de l’ammoniac au facteur 2 près :
V = 1/2 VNH3 = - 1/3 VH2
On doit considérer les deux cas qui suivent :
|
1.3 |
|
si le volume V varie (cas de réacteurs ouverts travaillant à pression constante) :
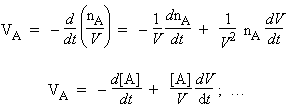
L’avancement et le degré d’avancement de la réaction
Soit la réaction générale : a A + b B ® + m M + n N . On définit l’avancement de la réaction l par la valeur du rapport du nombre de molécules de l’un des réactifs ou des produits sur son coefficient stœchiométrique qui ont disparu ou qui sont apparues après un certain temps :
l = - [(nA)0 - (nA)t]/a = - [(nB)0 - (nB)t]/b = [(nM)0 - (nM)t]/m . . .
(nA)0 représente le nombre de molécules du réactif A présentes dans le réacteur au temps t = 0 et (nA)t le même nombre observé au temps t = t. À nouveau le signe négatif est introduit pour les mêmes raisons invoquées plus haut.
La précédente définition de la vitesse spécifique n’aura pas la même grandeur puisqu’elle dépend du coefficient stœchiométrique qui affecte le produit ou le réactif choisi pour mesurer cette vitesse spécifique. Donc VA ¹ VB ... On sait, par contre, que :
VA = a/b VB ; . . . VM = - m/a VA
Pour rendre plus symétriques les expressions, on peut écrire :
![]()
dl est le degré d’avancement de la réaction; au temps t il est disparu l a moles de A, et il est apparu l m moles de M, etc. ... On peut aussi définir une vitesse du degré d’avancement de la réaction comme étant :
Vl = dl / dt
c- Représentation graphique
La représentation la plus fréquente, celle en fait qui correspond à la vitesse mesurée dans un réacteur à volume constant, montre la variation de la concentration d’un réactif ou d’un produit en fonction de la variable temps. Au temps t, l’expression de la vitesse n’est alors rien d’autre que la pente à la courbe :
[A] = ƒ(t), ou à la courbe [B] = ƒ(t), ...
d- Facteurs influençant la vitesse d’une réaction
Ils sont nombreux et, a priori, pas nécessairement évidents. On peut résumer cette dépendance de la façon symbolique suivante :
|
1.4 |
|
= k ƒ ([A], [B], ..., T, P, ...). |
On étudiera surtout l’effet de la température, T, de la pression, P, et, bien évidemment, des concentrations.
On verra plus loin l’influence de la température ainsi que celui de la pression. Le rapport entre la vitesse de réaction et la quantité de réactif a été proposé par deux scientifiques norvégiens en 1867. En effet, C. M. GULBERG (1836-1902) et P. WAAGE (1830-1900) ont établit que l’action chimique d’un composé est proportionnelle à sa concentration (nombre de moles par unité de volume). Cette relation est appelée le principe d’action de masse. Par exemple, en phase gazeuse, on sait maintenant que plus la concentration d’un réactif est grande et plus le nombre de collisions de ce réactif est grand. La possibilité de réaction chimique s’accroît donc avec la concentration.
3. L'ordre d'une réaction
Soit la réaction : a A + b B ® m M + n N + ... et supposons qu’expérimentalement on ait trouvé que la vitesse de la réaction pouvait se mettre sous la forme :
| 1.5 |
|
= k [A]a [B]b [M]m | avec k = f(T) |
On dira que :
a est l’ordre partiel de la réaction par rapport au réactif A,
ß est l’ordre partiel de la réaction par rapport au réactif B,
µ est l’ordre partiel de la réaction par rapport au produit M, etc.
En posant :
1.6 s = a + ß + µ + ...etc.
on dira aussi que s est l’ordre global de la réaction. Les ordres sont donc des notions purement expérimentales.
A- La règle de VAN’T HOFF : condition nécessaire d’une réaction élémentaire
Soit la réaction: a A + b B + ... ® produits. La règle de VAN’T HOFF s’écrit :
| 1.7 |
|
= k nA nA nA... × nB nB nB ... = k nAa nBb ... |
| a fois b fois |
La condition de VAN’T HOFF
traduit que puisque la réaction se passe dans une étape élémentaire, il faut
nécessairement que tous les réactifs se rencontrent en même temps en un point de
l’espace. La probabilité de trouver tous les réactifs en un point de l’espace est
proportionnelle à la concentration de chacun des réactifs. Si i molécules
d’un
produit quelconque sont nécessaires, alors la concentration de ce réactif intervient à
la puissance i. Il s’ensuit que les ordres partiels de la réaction sont égaux
aux coefficients stœchiométriques de la réaction; l’ordre global est égal à leur
somme: c’est donc un entier positif, et simple : 1, 2 3,... On voit aussi que les
concentrations des produits n’interviennent pas dans l’expression de la vitesse. Si cela
était le cas, la réaction ne serait certes pas une réaction élémentaire.
Par exemple :
H2 + I2
® 2
HI
vH2 = k [H2]1
[I2 ]1 et
s = 2.
Dans cette équation l’ordre global est égal à la molécularité.
B. La règle de VAN’T HOFF n’est pas suffisanteUn exemple en fera une démonstration fort simple. Soit donc une réaction procédant à travers deux étapes élémentaires successives :
| A + B |
|
C | équilibre rapide |
| C + D | ® | produits | réaction lente |
|
Globalement : |
|||
| A + B + D | ® | produits | réaction globale |
Bien sûr, c’est la réaction la plus lente qui va imposer sa vitesse à l’ensemble de la réaction globale, de telle sorte que, en vertu de la condition nécessaire de VAN’T HOFF :
v = k [C] [D]
En outre, la constante thermodynamique de l’équilibre, KT, est telle que :
![]()
La concentration de l’espèce intermédiaire C, [C], n’est en général pas connue. En remplaçant dans l’équation précédente, il vient :
v = k KT [A] [B] [D]
Dans ce cas l’ordre global est bien identique à la molécularité, les ordres partiels sont bien égaux aux coefficients stœchiométriques de la réaction et pourtant la réaction n’est pas une étape élémentaire. La règle de VAN’T HOFF n’est donc pas une règle suffisante.
C. L’ordre est simple mais différent de la stœchiométrie
La réaction ne se déroule pas en une seule étape et la réaction globale est constituée d’une suite de réactions élémentaires dont l’une (la plus lente) impose sa vitesse.
Par exemple: 2 NO• + 2 H2 ® 2 H2O + N2
Dans cette équation • représente un électron non apparié ou encore célibataire. L’expression expérimentale de la vitesse devient : v = kexp [NO•]2 [H2 ]
L’ordre est 3 et la stœchiométrie est de 4. La condition de VAN’T HOFF n’est pas satisfaite. La réaction n’est donc pas une étape élémentaire. Quelles sont les possibilités de mécanismes? Essayons le suivant :
2 NO• + H2 ® H2O2 + N2 réaction lente, suivie de :
H2O2 + H2 ® 2 H2O réaction rapide
La réaction lente impose sa vitesse. Donc : v = k [NO·]2 [H2]. Mais on peut aussi imaginer, proposer, un autre mécanisme :
| 2 NO |
|
N2O2 | équilibre rapide, KT |
| N2O2 + H2 | ® | H2O2 + N2 | réaction lente |
| H2O2 + H2 | ® | 2 H2O | réaction rapide |
v = k2 [N2O2] [H2] Or, KT = [N2O2] /[NO·]2
Donc, v = k KT [NO·]2 [H2]. Cet exemple montre que l’équation de vitesse ne permet pas toujours d’établir avec certitude l’exact mécanisme réactionnel. Le chimiste devra user d’imagination pour déterminer lequel des deux mécanismes est le bon (ou peut-être est-ce un troisième ?).
D. L’ordre n’est pas simple
Dans ce cas, l’ordre peut être positif, négatif, fractionnaire, ou même extrêmement compliqué. La réaction est nécessairement une réaction complexe, c’est-à-dire qu’elle ne peut progresser qu’à travers une suite plus ou moins complexe d’étapes élémentaires.
Par exemple, pour la réaction H2 + Br2 ® 2 HBr,
on montre expérimentalement que la vitesse de la réaction s’écrit sous la forme :
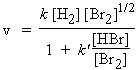
Que peut-on dire de l’ordre global ? Des ordres partiels ? La réaction est assurément complexe puisqu’au moins l’un des produits intervient dans l’expression de la vitesse. On traitera ce cas plus en détail plus loin.
E.
ORDRE ET MOLÉCULARITÉ
Il
n’existe pas nécessairement une adéquation entre l’ordre et la molécularité.
On l’a vu plus haut, la molécularité est égale aux nombres de
particules, d’individus chimiques (molécules, radicaux, ions,…) qui
participent à la réalisation d’une étape élémentaire. L’ordre,
au contraire, est une notion expérimentale qui traduit certaines caractéristiques
du mécanisme réactionnel, mécanisme qui peut comprendre plusieurs étapes élémentaires.
S’il y a parfaite adéquation entre la molécularité et l’ordre
d’une réaction, c’est accidentel.
tert-C4H9OH ® C4H8 + H2O ordre 1, molécularité 2 ;
CH3OCH3 +
I• ®
CH3COCH2I + H•
ordre 0,
molécularité 2
2
NO• +
O2
®
2 NO2
ordre
3, molécularité 2
4. Principes guidant le choix d’un mécanisme réactionnel
A. Il faut maintenir la plus grande simplicité dans les réactions élémentaires. En d’autres termes, la nature n’affectionne pas particulièrement ce qui est compliqué. C'est le principe de moindre changement des structures. À priori, il faut se limiter à des réactions élémentaires d’ordre inférieur ou égal à 2, sauf dans de rares cas où il pourra être égal à 3. On verra ces cas ultérieurement.
B. Bien entendu, les lois de la thermodynamique doivent être respectées. En conséquence, il faut éviter les réactions endothermiques, à moins que l’on dispose de l’énergie suffisante.
C. Enfin, retour au principe de simplicité, il ne faut admettre que des changements très simples de structures. L’exemple des combustions d’hydrocarbures est suffisamment explicite. Cette réaction réclame la rupture de 10 liaisons C¾H, 3 liaisons C¾C et 13/2 liaisons O=O ! Voilà tout un programme qu’on ne saurait réaliser en une seule étape, même avec les meilleurs outils ou bonnes intentions,...
n-C4H10 + 13/2 O2 ® 4 CO2 + 5 H2O.
5. Nature des entités participant aux actes élémentaires
Il y en a de plusieurs types ou nature. Toute entité chimique (et quel produit ne l’est pas ?) est susceptible d’intervenir. Mentionnons toutefois :
a- Les espèces elles-mêmes, normales ou activées.
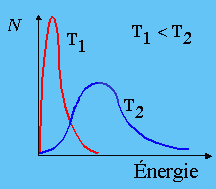
On sait qu’à une température T, les molécules supportent une énergie dite thermique qui est distribuée dans les niveaux d’énergie de translation, de rotation, de vibration. Le contenu de cette énergie n’est pas fixe. Comme il y a échange d’énergie entre les molécules à travers les collisions, l’énergie moyenne supportée par les molécules ressemble à une distribution de type gaussien. Plus la température augmente et plus les molécules sont énergiquement chaudes. Si la majorité des molécules ont une énergie moyenne relativement faible, une minorité peut avoir une énergie interne très importante. Celles qui auront une énergie interne supérieure à l’énergie minimum correspondant au seuil réactionnel, ces molécules seront susceptibles de réagir, Fig. 1.1.
Par exemple :
| CF3OOCF3 | ® | 2 CF3O•, réaction qui a lieu vers 450 °C, suivie de |
| CF3O• | ® | F• + CF2O |
| F• + CF3OOCF3 | ® | CF3OF + CF3O· ... |
| CH3N=NCH3 | ® | C2H6 + N2, réaction se faisant vers 450 °C. |
 |
 |
| Distribution thermique de l’énergie | |
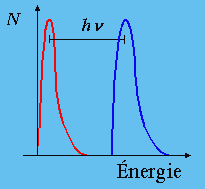
| Figure 1.1. par absorption de chaleur. | Figure 1.2. par absorption de lumière.. |
Dans ces deux cas, l’énergie est transmise aux molécules à travers un faisceau de photons ou de toute autre particule. Ce peut être des électrons, comme dans le cas de la spectrométrie de masse, des protons, des rayons a, b, ou g (Fig. 1.2). Par exemple :
|
|
+ h n ® H• + •CH3 + (CH2=C=CH2 ou CH3-CºCH) |
Cette réaction a un rendement important lorsque la longueur d’onde l est de 140 nm. Dans le proche UV, la réaction d’isomérisation du trans-2-butène vers la formation du cis-2-butène est aussi bien connue. Un autre exemple très concret est celui de la synthèse chlorophyllienne. Dans ce cas, l’énergie solaire est emmagasinée par la chlorophylle et la plante l’utilise pour synthétiser des carbohydrates à partir du gaz carbonique atmosphérique. Bien entendu, dans chacun de ces cas, il faut que la molécule puisse absorber (ne soit pas transparente) le rayonnement incident. On verra plus tard les techniques employées.
Dans ce cas l’énergie est apportée par l’intermédiaire d’un courant électrique. L’exemple le mieux connu dans la région du Saguenay est la production de l’aluminium à partir de la bauxite.
b. Les formes réactives
Elles sont variées, et l’on verra en temps opportun des cas très concrets. On verra que
pour réagir, le plus souvent, une molécule doit atteindre une configuration
particulière, que l’on appellera pour le moment géométrique. Un exemple suffira à en
indiquer l’importance. Par exemple, il est bien connu que les cétones réagissent avec le
brome à travers leur forme énolisée :
|
|
processus lent |
réaction suivie de:
R-C(OH)=CH2
+ Br ®
R-C(OH)Br-CH2Br
®
R-C(=O)-CH2Br
+ HBr.
Ces deux dernières réactions sont, elles, très rapides.
c. Les formes intermédiaires actives
On peut imaginer toutes sortes de formes intermédiaires réactives. Par exemple :
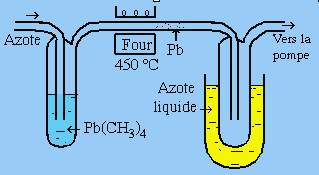
Il est important de mentionner ici les expériences du miroir de PANETH (1924) de même que du dispositif un peu plus sophistiqué de POLANYI (1933) (Fig. 1.3 et 1.4).
Pb(CH3)4 + chaleur ® Pb + 2 C2H6
|
|
Figure 1.3. L’expérience de PANETH.
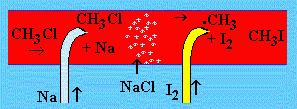
Dans le cas présent, le plomb tétraméthyle est entraîné par un courant d’azote qui barbote dans le liquide. Il passe dans un tube en quartz chauffé à 450 °C. À la sortie du four il apparaît un film métallique (miroir) qui ne peut être que du plomb. On doit donc imaginer que les liens Pb-CH3 ont été rompus et que les espèces radicalaires ·CH3 se sont recombinées pour former l’éthane. La preuve de la présence (formation) des radicaux est indirecte. Dans l’expérience de PANETH, la mise en contact de vapeur de sodium avec chlorure de méthyle fait apparaître des cristaux blancs qui se révèlent être du chlorure de sodium. La réaction doit laisser des fragments méthyles. Ceux-ci, avant qu’ils n’aient le temps de se combiner entre eux pour former l’éthane comme dans le cas précédent, sont mis en présence d’iode. Il se forme alors de l’iodure de méthyle, prouvant ainsi la formation intermédiaire de radicaux méthyles.
|
|
Figure 1.4. Dispositif de POLANYI.
On retrouve ici tout le domaine de l’électrochimie, des réactions en solutions, plus précisément dans des solvants fortement polaires comme l’eau. Les exemples sont nombreux et l’étudiant en a déjà une idée suffisante. On en verra d’autres exemples de façon plus approfondie dans les chapitres qui suivent.
6. Influence de la température sur la vitesse de réaction
A- La constante de vitesse
On a vu que d’une façon générale, (équation 1.5) : v = kT ƒ([A]; [B],...), ou, quand il y a un ordre :
v = kT ƒ ([A]a, [B]ß,...).
La constante kT ne dépend ni du temps, ni des concentrations, mais uniquement de la température T. kT est appelé la constante de vitesse de la réaction. La IUPAC (Union internationale de chimie pure et appliquée) recommande l’appellation de coefficient de vitesse.
B- L’équation d’ARRHÉNIUS
(1889)
ARRHÉNIUS a montré que la constante peut se mettre sous la forme :
C'est l'équation d'ARRHÉNIUS
| 1.8 | Ln kT = B - C/T |
| Note : |
|
est semblable à |
|
équation mieux connue sous le nom de loi de VAN’T HOFF. Graphiquement, au lieu de représenter la variation de kT en fonction de la température, Fig. 1.5, on préfère plutôt représenter le Ln (ou le log) de la constante de vitesse en fonction de 1/T(K), Fig. 1.6. Il est à remarquer que l’équation d’ARRHÉNIUS ne prévoit aucune dépendance de la température sur le facteur pré exponentiel.
 |
 |
Figures 1.5. et 1.6. Représentations de kT en fonction de la température.
Exemple :
La constante de vitesse de la réaction bimoléculaire suivante a été mesurée à plusieurs températures.
NO + ClNO2 ® NO2 + ClNO
Les résultats obtenus sont les suivants :
|
T (K) |
300 |
311 |
323 |
334 |
344 |
|
10-7k/cm3mol-1s-1 |
0,79 |
1,25 |
1,64 |
2,56 |
3,4 |
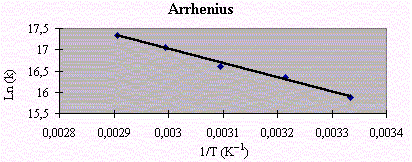
La valeur de l’énergie d’activation :
|
|
= - 3 377,2 K |
Ea = 28,1 kJ · mol-1
tandis que le facteur pré exponentiel : Ln A = 27,15 ou encore
A = 6,1 1011 cm3 · mol-1 · s-1
C- L’énergie d’activation
La formule 1.8 peut encore s’écrire sous la forme : kT = A e- C/T et de façon plus explicite :
| 1.9 |
kT = A e-Ea/R T |
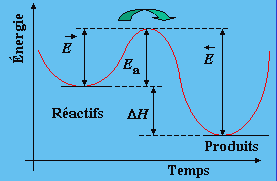
Le terme Ea est appelé l’énergie d’activation de la réaction et A est le facteur pré exponentiel. C’est l’équivalent d’une barrière de potentiel qui s’oppose à l’avancement de la réaction. Graphiquement, cette barrière se représente selon la figure 1.7 qu’on nomme un diagramme des coordonnées de la réaction.
|
|
Figure 1.7. Diagramme des coordonnées de la réaction.
Soit la réaction :
|
a A + b B |
m M + n N |
Selon le graphique précédent, il est facile de voir que
![]()
représentent respectivement les énergies d’activation des réactions directe et inverse. À l’équilibre thermodynamique, on peut écrire :
| 1.10 |
|
= KT |
La loi de VAN’T HOFF s’exprime de la manière suivante :
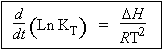
ou encore
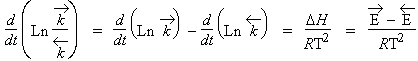
| D’où 1.11 |
|
Þ Ln k = b - E/RT ou k = A e-Ea/RT |
La grandeur Ea est l’énergie d’activation de la réaction; A est la constante absolue de la réaction, encore mieux appelé le facteur pré exponentiel.
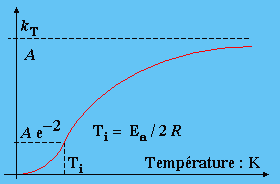
D- L’influence de la température
Soit donc à établir les variations de kT avec la température : kT = A e- Ea/T.
Lorsque la température T croît, le facteur Ea/RT décroît, eEa/RT décroît également et 1/eEa/RT , comme e-Ea/RT, croît k est donc une fonction croissante de la température. Aux limites, la fonction ne devient pas infinie. Lorsque T ® 0, Ea ® , et 1/eEa/RT = e-Ea/RT ® 0 . Quand, au contraire, T ® ¥ , eEa/RT ® 1, tout comme 1/eEa/RT. Dans ce cas, kT tend donc vers la valeur de A. La courbe kT = ƒ(T) admet donc une asymptote lorsque la température tend vers l’infini (Fig. 1.8). Soit I le point d’inflexion. Les coordonnées de ce point I peuvent être déterminées. En effet, en ce point I, la dérivée seconde
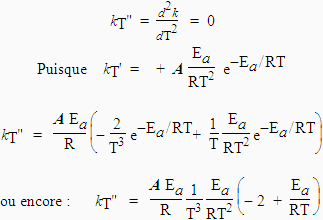
kT" = 0 si Ea/R T = 2; ou encore si T = Ea /2 R .
En général, l’énergie d’activation des réactions chimiques est telle que 40 < Ea < 400 kJ·mol-1. Compte tenu de la valeur de R, la température à laquelle apparaît le point d’inflexion, Ti, est telle que : 2 500 < Ti < 25 000 K. Cette température est, à toutes fins pratiques, en dehors des limites accessibles, de telle sorte qu’expérimentalement on demeure dans la zone de température inférieure à cette valeur de Ti. On peut aussi, calculer la valeur de k en ce point d’inflexion :
ki = A e-Ea/R 2R/Ea = A e-2 en se rappelant que, en général, k < ki.
Figure 1.8. Variations de kT avec la température.
7. Choix d'un mécanisme réactionnel
C’est bien évidemment établir la succession des réactions ou étapes élémentaires d’un mécanisme global. Il faudra :
mathématique de l’expression de la vitesse;
On voit donc que la cinétique chimique est fondamentalement une science expérimentale. Cela n’empêche pas qu’elle est susceptible d’une théorisation très avancée.
a. Méthodes expérimentales de mesures de vitesse de réactions
Généralités, classement, méthodes, ...
On a mentionné l’importance de la température : celle-ci a une influence déterminante sur le facteur pré exponentiel. Il est donc impératif, si l’on veut mesurer une vitesse de réaction, de travailler à température bien établie. On utilisera donc des systèmes expérimentaux thermostatés. En deuxième lieu, on a vu que, pour mesurer la vitesse d’une réaction chimique, il suffit de suivre la diminution de la concentration d’un réactif ou l’augmentation de celle d’un produit. Il suffira donc, par exemple, de suivre la variation d’une propriété clairement associée à l’un des réactifs ou des produits. Si l’on peut suivre l’ensemble des produits et des réactifs, on sera bien sûr plus assuré de la vitesse de réaction sinon du mécanisme.
On peut classer les méthodes expérimentales de diverses manières. Notons :
c- Quelques méthodes physiques de mesure des vitesses de réaction
Note : compte tenu de la disponibilité de plusieurs monographies relatives aux systèmes et montages expérimentaux, on se contentera ici de présenter quelques cas à peine détaillés tout en incitant le lecteur à consulter ces monographies (voir à la fin de ce chapitre: Pour en savoir plus).
A ® m M + n N
On peut établir les bilans suivants :
| Bilans : | A | M | N | total |
|---|---|---|---|---|
| - avant | P0 | 0 | 0 | P0 |
| - après | P0 - a P0 | m a P0 | n a P0 | [(1-a) + a(m+n)]P0 = P |
| - à t = t¥ | 0 | m P0 | n P0 | (m + n)P0 = P¥ |
On peut réécrire la ligne médiane du tableau en remarquant que a est la proportion du réactif A qui a disparu et si P est la pression totale au temps t :
P / P0 = 1 + a(m + n - 1)
Au temps infini, t¥ , la pression est devenue P¥ : P¥ = (m + n) Po et en combinant les deux dernières équation, il vient :
|
ou encore |
|
Soit à étudier la réaction d’hydrolyse d’un ester en milieu acide.
|
CH3COOCH3 + H2O + H+ |
|
CH3COOH + CH3OH + H+ |
Cette réaction (équilibre) peut être suivie en fonction du temps simplement par un titrage de l’acide acétique formé par une base. Cette méthode se rattache aux méthodes destructives ou discontinues.
L’hydrolyse d’un acétal en milieu acide donne lieu pendant la réaction au remplacement d’une molécule d’acétal par trois molécules : une molécule d’aldéhyde et deux molécules d’alcool.
CH3CH(OC2H5)2 + H2O + H+ ® CH3CHO + 2 C2H5OH + H+
Un judicieux montage d’un tube capillaire au-dessus du ballon réactionnel permet de visualiser l’augmentation de volume faible mais mesurable en fonction du temps.
La mesure d’augmentation de masse est une technique particulièrement adaptée aux mesures d’oxydation des métaux.
Cu(solide) + 1/2 O2(gaz) ® CuO(solide)
Un cas simple permet de visualiser la modification de conductivité électrique. Soit la réaction d’hydrolyse d’un ester par des ions hydroxyles. L’équation chimique montre, qu’au cours de la réaction, les ions hydroxyles, OH-, sont remplacés par des ions acétates beaucoup moins mobiles. La conductivité de la solution diminue donc au fur et à mesure des progrès de la réaction.
CH3COOC2H5 + OH- ® CH3COO- + C2H5OH
Ces techniques sont prisées car le plus souvent elles s’intègrent dans des systèmes de mesures continues et non-destructives. La bromuration de l’acétone peut être suivie par spectroscopie d’absorption dans le visible. On peut en effet mesurer en fonction du temps la disparition du brome qui est le seul produit coloré apparaissant dans la réaction suivante :
CH3COCH3 + Br2 ® CH3COCH2Br + Br - + H+
Des techniques récentes permettent de suivre l’évolution de certains produits intermédiaires de réaction dans des temps de l’ordre de 100 femtosecondes en utilisant des lasers ultra-rapides. On a ainsi pu mettre en évidence que la réaction de DIELS-ALDER (addition de l’éthylène sur le cyclopentadiène) procède selon deux chemins ayant approximativement la même barrière de potentiel.
Un exemple bien connu est celui de la variation du pouvoir rotatoire observé dans la réaction d’inversion du sucrose (sucrose ® glucose + fructose) :
C12H22O11 + H2O + H+ ® C6H12O6 + C6H12O6 + H+
Le sucrose est dextrogyre alors que la somme (glucose + fructose) est légèrement lévogyre. Cette réaction peut aussi être suivie à l’aide d’un colorimètre.
Ces méthodes se rattachent aux méthodes discontinues et destructrices. Elles sont d’une grande utilité lorsque l’on veut mesurer non pas des vitesses absolues de réaction mais plutôt des vitesses relatives. Soit par exemple les réactions entre un radical X et deux additifs. Si l’on peut mesurer les produits issus de chacune des deux réactions, on saura mesurer le rapport des deux constantes de vitesse. Si de surcroît, la valeur absolue de l’une des deux constantes est connue, la détermination de la valeur absolue de la seconde est immédiate.
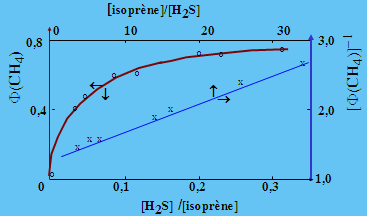
La photolyse de l’isoprène vers 174,4 nm en phase gazeuse produit des radicaux méthyles avec un rendement quantique (on verra plus loin la définition de cette grandeur) important, de l’ordre de 0,60. Ces radicaux libres réagissent probablement par addition sur l’isoprène. En ajoutant de petites quantités d’hydrogène sulfuré, on introduit une réaction compétitive : il y a formation de méthane. Le mécanisme est le suivant (Demers, R. et al., Can. J. Chem., 56, 1864 (1978)):
[1] CH3• + H2S ® CH4 + HS•
[2] CH3• + C5H8 ® C6H11•
Un traitement cinétique simple dont verra aussi plus loin le rationnel permet de lier le rendement quantique du méthane aux concentrations d’isoprène et d’hydrogène sulfuré. La figure 1.9 permet d’apprécier la valeur du résultat et donc de l’approche qui consiste à mesurer le méthane par chromatographie en phase gazeuse.
[F(CH4)]-1 = [F(CH3•)]-1 + [F(CH3•)]-1 k2 /k1 [isoprène]/[H2S]
|
|
Figure 1.9. Variation du rendement
en méthane et de son
inverse dans la photolyse du mélange isoprène - hydrogène sulfuré.
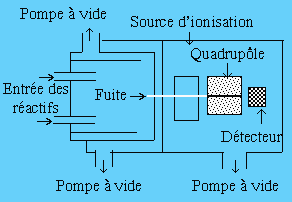
Le spectromètre de masse est un appareil quasi-universel de mesure d’entités chimiques, malheureusement surtout limité à la phase gazeuse. Néanmoins les techniques en général et du vide en particulier ayant tellement évoluées, c’est un appareil très versatile. La figure 1.10 illustre le principe de son fonctionnement. Le réacteur, alimenté en réactif, opère en mode dynamique (flux continu). Au fond du réacteur un minuscule orifice (@ 0,4 mm2) permet la fuite du mélange réactionnel. Un deuxième orifice de même grandeur est situé en face le précédent : la fuite se transforme en un faisceau moléculaire qui s’introduit dans les éléments d’un système quadripolaire. Celui-ci fait office de sélecteur de masse. Finalement le détecteur d’ions permet de mesurer les entités chimiques. Le système de réactions des radicaux libres éthyles avec l’oxygène moléculaire a ainsi été étudié par MM. Plumb et Ryan en 1981. Ils ont pu mesurer à la fois les radicaux éthyles et l’éthylène permettant d’établir à 11 % la part relative de la formation de l’éthylène :
C2H5• + O2 ® C2H5O2• et C2H5• + O2 ® C2H4 + HO2•
|
|
Figure 1.10. Schéma de montage d’un dispositif de
mesure de cinétique de réaction
par spectrométrie de masse.
Soit par exemple la réaction d’hydrolyse d’un ester par les ions hydroxyles :
CH3COOC2H5 + OH- ® CH3COO- + C2H5OH
Au cours de la réaction les ions OH- sont remplacés par des ions acétates. Ces derniers ayant une conductivité électrique plus faible que celle des ions hydroxyles, on peut donc mesurer en fonction du temps la diminution de cette conductivité électrique.
Ces méthodes s’appliquent avec plus ou moins de succès selon que la réaction est en phase gazeuse, liquide ou solide. La température est aussi un facteur à considérer. Le caractère ouvert ou fermé du réacteur peut être déterminant. Finalement, la vitesse de réaction a aussi une influence déterminante sur la méthode de mesure. Une réaction qui se réalise en une microseconde s’accommode mal d’un chronomètre gradué en secondes. Il faudra donc disposer d’un appareil qui pourra suivre une propriété physique ou chimique chaque dixième, voire chaque centième de microseconde. Actuellement, les appareils les plus sophistiqués peuvent suivre une réaction dans des temps aussi courts que 10 picosecondes, ou encore en 10-11 secondes. On verra plus loin des méthodes modernes de mesure de réactions extrêmement rapides.
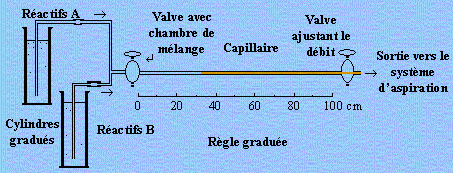
En résumé, l’imagination du chimiste, son flair et une connaissance intime des mécanismes réactionnels, sont souvent plus importants que les essais de classification qui gardent néanmoins leurs valeurs intrinsèques. L’introduction des techniques de flux continu est un exemple de cette imagination qui permet d’en visualiser les multiples facettes. L’oxydation du bisulfite de sodium par l’eau oxygénée en présence d’iodure de potassium et d’amidon est une réaction relativement rapide. Deux solutions, l’une contenant de l’eau oxygénée en milieu HCl (réactifs A), l’autre le bisulfite, l’iodure et l’amidon (réactifs B) sont physiquement séparées dans deux réservoirs identiques (Fig. 1.11). Le système permet l’introduction simultanée et aux mêmes débits de ces deux solutions dans une chambre de mélange. Celle-ci est prolongée par un capillaire lui même attaché à un système de succion. Dépendant du débit du mélange réactionnel dans le capillaire on pourra mesurer la distance à laquelle apparaît la couleur attendue. En établissant les liens appropriés entre la distance d’apparition de la couleur, le débit dans le capillaire, on saura mesurer des vitesses réactionnelles de l’ordre de la seconde et moins.
|
|
Figure 1.11. Principe du système de mesure en flux continu.
8. Conclusions
|
9. Exercices
1- Expérimentalement, la réaction 2 NO + Cl2 ® 2 NOCl donne les résultats suivants : en doublant la concentration des deux réactifs la vitesse v est multipliée par huit (8); en doublant seulement la concentration du chlore, on double seulement la vitesse. Quels sont les ordres partiels et global de la réaction?
2- La réaction I
- + OCl - ® Cl - + OI - suit une loi de vitesse telle que vexp = k' [I -] [OCl -], mais k' se révèle une fonction de la concentration des ions OH -. À 25 °C, pour des concentrations d’ions OH - de 1,00 M, 0,5 M et 0,25 M, k' est respectivement égal à 61, 120 et 230 litre/(mol·s). Quel est l’ordre de la réaction par rapport aux ions hydroxyles ?Est-ce que le mécanisme proposé ci-après est conforme à cette loi de vitesse? Prouvez votre affirmation.
| OCl- + H2O |
|
HOCl + OH- | équilibre rapide |
| HOCl + I- | ® | HOI + Cl- | réaction lente |
| HOI + OH- | ® | H2O + OI- | réaction rapide |
3- Le nitramide O2NNH2 se décompose lentement en solution aqueuse d’après la réaction globale suivante :
O2NNH2 ® N2O + H2O
avec la loi expérimentale de vitesse :
![]()
Lequel des trois mécanismes, A, B ou C, suivants semble le plus approprié ?
| A- | [1] | O2NNH2 |
|
N2O + H2O | réaction lente |
| B-
|
[2] | O2NNH2 + H+ |
|
O2NNH3+ | équilibre rapide |
| [3] | O2NNH3+ |
|
N2O + H3O+ | étape lente | |
| C-
|
[4] | O2NNH2 |
|
O2NNH- + H+ | équilibre rapide |
| [5] | O2NNH- |
|
N2O + OH- | étape lente | |
| [6] | OH- + H+ |
|
H2O | réaction rapide |
Quelle est la relation entre le k de la loi expérimentale et celui de la loi cinétique expérimentale et les constantes des vitesses des réactions du mécanisme choisi.
4- En solution acide, la vitesse de la réaction :
NH4+ + HNO2 ® N2 + 2 H2O + H+
est compatible avec le mécanisme
| HNO2 + H+ |
|
H2O + NO+ | équilibre rapide |
| NH4+ |
|
NH3 + H+ | équilibre rapide |
| NO+ + NH3 | ® | NH3NO+ | étape lente |
| NH3NO+ | ® | H2O + H+ + N2 | réaction rapide |
Écrire la loi cinétique qui est compatible avec le mécanisme en exprimant :
|
|
= ƒ ([NH4+]; [HNO2]; [H+]) |
5- La racémisation du pinène en phase gazeuse a une vitesse de réaction qui est égale à 2,2 10-5 min-1 à 457,6 K et à 3,07 10-3 à 510,1 K. Calculez l’énergie d’activation de la réaction.
Réponse : 43 600 cal·mol-1 = 182,25 kJ·mol-1
6- On suit la décomposition du pentoxyde d’azote, N2O5, en fonction du temps à différentes températures.
| T (°C) | 0 | 25 | 35 | 45 | 55 | 65 |
| k (s-1) | 7,87 10-7 | 3,46 10-5 | 1,35 10-4 | 4,98 10-4 | 1,50 10-3 | 4,87 10-3 |
Déterminer graphiquement l’énergie d’activation de la réaction et trouver graphiquement la constante de vitesse à 50 °C.
7- La constante de vitesse spécifique de la réaction entre le nitropropane et les ions OH- en solution est donnée par la relation :
log k = - (3 163 / t ) + 11,90 où le temps t est exprimé en minutes et les concentrations en mole/litre.
On indique en outre que la réaction est d’ordre 2. Indiquez en quelles unités s’exprime la constante de vitesse k. Calculez l’énergie d’activation à 10 °C.
Réponse : 14,57 kcal·mol-1
8- L’énergie d’activation pour une réaction de décomposition d’une molécule quelconque est de 83 600 J·mol-1. Trouvez la fraction des molécules qui ont l’énergie suffisante pour franchir la barrière de potentiel de la réaction à 27 °C et à 227 °C.
Réponse: P27 °C = e-33,3
9- La constante de vitesse d’une réaction chimique est telle que :
log k (s-1) = 14,21 - 112,4/q où q = 2,303 R T kJ·mol-1
Déterminez l’énergie d’activation de la réaction en calories·mol-1.
10- Soit la réaction:
CH3CH(CH3)CH2CH2Cl ® CH3CH(CH3)CH=CH2 + HCl
Elle donne les résultats suivants :
| temp., (°C) | 420,1 | 430,1 | 440,0 | 444,5 | 450,2 | 460,1 | 470,0 |
| 104 k, (s-1) | 2,45 | 4,27 | 7,34 | 10,08 | 12,94 | 22,15 | 38,04 |
Calculez l’énergie d’activation et le facteur pré exponentiel en estimant l’incertitude expérimentale.
Pour en savoir plus :
Alain Dumon, La naissance des concepts de la cinétique chimique au XIXe siècle, L'atualité chimique, Société chimique de France, N° 436, 41-45 (janvier 2019).
Shoemaker, D. P., C. W. Garland et J. I. Steinfeld, Experiments in Physical Chemistry, 3e édition, McGraw-Hill Book Company, New York, 1974; QD457.S559.
Drago, R. S. et J. I. Steinfeld, Experiments in General Chemistry, 3e édition, Allyn and Bacon inc., Boston, 1970.
Daniels, F., R. A. Alberty, J. W. Williams, C. D. Cornwell, P. Bender et J. E. Harriman, Experimental Physical Chemistry, 7e édition, McGraw-Hill Book Company, New York, 1970; QD 457 D187.
Roubi, M., Femtoseconds Dynamics Reveal Two Paths in Diels-Alder Reaction, Chemical & Engineering News, 34, 7 octobre 1996.
Sur le NET :
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes industries, les Fondations, ...
Dernière mise à jour : 2021-07-01.
x x x x x x x x x x x x x x

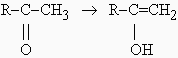
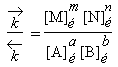
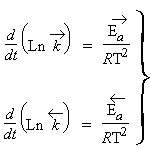
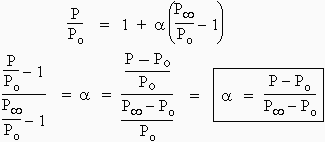
{kind=link}